
This AOP is open for adoption and licensed under the BY-SA license. The BY-SA license allows reusers to distribute, remix, adapt, and build upon the material in any medium or format, so long as attribution is given to the creator. The license allows for commercial use. If you remix, adapt, or build upon the material, you must license the modified material under identical terms.
AOP: 519
Title
Cardiac ion channels blockade leading to increased incidence of cardiovascular morbidity and mortality
Short name
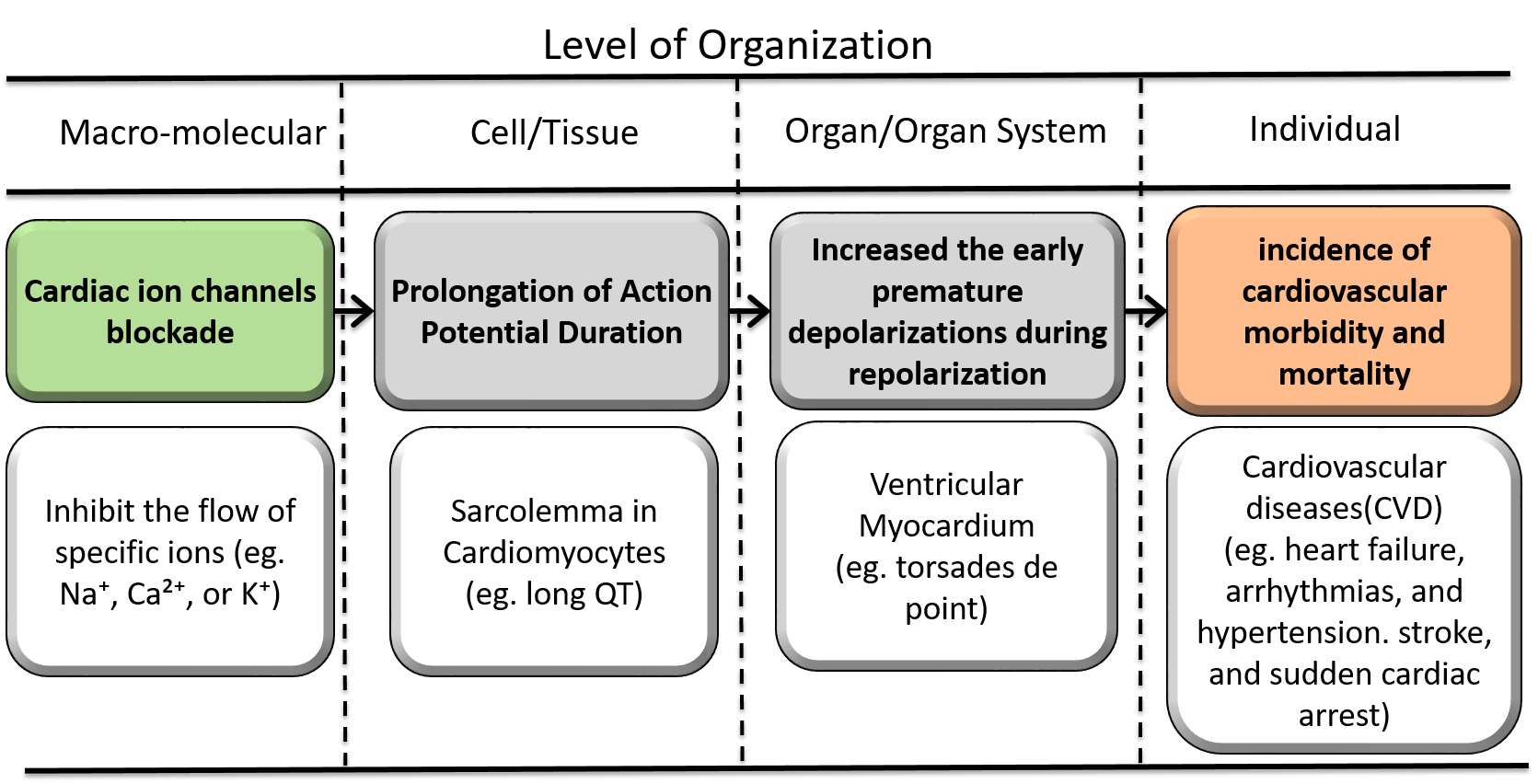
Graphical Representation
Additional AOP Exploration Options
Click links below to explore AOP 519, Cardiac ion channels blockade leading to increased incidence of cardiovascular morbidity and mortality in tools offered by third parties.
Point of Contact
Contributors
- Young Jun Kim
Coaches
OECD Information Table
| OECD Project # | OECD Status | Reviewer's Reports | Journal-format Article | OECD iLibrary Published Version |
|---|---|---|---|---|
This AOP was last modified on July 03, 2026 04:23
Revision dates for related pages
| Page | Revision Date/Time |
|---|---|
| Cardiac ion channels blockade | November 21, 2024 05:28 |
| Prolongation of Action Potential Duration | January 29, 2023 11:26 |
| Increased the early premature depolarizations during repolarization | November 26, 2024 12:57 |
| Increased incidence of cardiovascular morbidity and mortality in the general population | August 21, 2021 11:21 |
| Cardiac ion channels (e.g., potassium, sodium, calcium) leads to Prolongation of Action Potential | November 21, 2024 05:32 |
| Prolongation of Action Potential leads to Early premature depolarizations | November 21, 2024 05:33 |
| Early premature depolarizations leads to Increased incidence of cardiovascular morbidity and mortality | November 21, 2024 05:33 |
| Cisapride | December 13, 2021 06:56 |
| Terfenadine | December 13, 2021 06:55 |
| Arsenic | April 27, 2021 00:15 |
| Halogenated Hydrocarbons | November 21, 2024 05:14 |
| terfenadine | November 21, 2024 05:16 |
Abstract
This Adverse Outcome Pathway (AOP) describes the mechanistic sequence of events linking cardiac ion channel blockade to increased incidence of cardiovascular morbidity and mortality . The Molecular Initiating Event (MIE) involves the blockade of key cardiac ion channels, such as hERG potassium channels, which disrupt ion currents critical for maintaining normal cardiac action potential. This triggers a cascade of Key Events (KEs), beginning with the prolongation of action potential duration (APD), which promotes early afterdepolarizations (EADs). These EADs lead to triggered activity, resulting in the onset of cardiac arrhythmias, such as Torsades de Pointes or ventricular fibrillation. Sustained arrhythmias impair cardiac output, culminating in sudden cardiac death. The Key Event Relationships (KERs) demonstrate strong biological plausibility and are supported by empirical evidence, including dose-response relationships, in vitro and in vivo studies, and clinical data linking QT prolongation to arrhythmic risk ( e.g.,Regulatory guidelines e.g., ICH S7B, E14). Despite some uncertainties, such as individual variability in repolarization reserve, this AOP provides a robust framework for predicting the cardiotoxicity of compounds, guiding risk assessment, drug development, and regulatory decision-making.
AOP Development Strategy
Context
Sudden cardiac death caused by arrhythmias is a major concern in cardiotoxicity, especially in the context of pharmaceutical development and chemical safety assessment. Many drugs and chemicals can inadvertently block cardiac ion channels, such as the hERG potassium channel, leading to arrhythmogenic events. The ability to predict and mitigate such risks early in the development process is critical for ensuring patient safety and regulatory compliance.
This AOP, "Cardiac Ion Channel Blockade Leading to increased incidence of cardiovascular morbidity and mortality." provides a mechanistic understanding of how ion channel blockade initiates a series of cellular and tissue-level events, ultimately resulting in a fatal adverse outcome. By defining the sequence of molecular and physiological changes and their relationships, the AOP serves as a structured framework to guide the evaluation of cardiotoxic risks. It is particularly relevant in areas such as drug screening, environmental toxicology, and the development of computational models for safety assessment. The application of this AOP extends to identifying compounds with ion channel blocking properties, designing safer drugs, and supporting regulatory decision-making by incorporating mechanistic data into risk assessment frameworks. This approach aligns with the principles of evidence-based toxicology, offering a pathway to more predictive and reliable safety assessments.
Strategy
1. Problem Formulation
Define the Scope: Establish the biological and regulatory significance of cardiac ion channel blockade in causing arrhythmias and sudden cardiac death.
Biological Scope: Focus on ion channels critical for cardiac electrophysiology (e.g., hERG, sodium, and calcium channels).
Regulatory Scope: Align with safety evaluation needs in pharmacology and toxicology, such as drug-induced QT prolongation, environmental toxicants, and cardiac risk assessments.
End Users: Identify the primary users of the AOP, including regulatory agencies (e.g., FDA, EMA), pharmaceutical developers, and environmental toxicologists.
2. Literature Review and Data Collection
Identify Key Events (KEs):
Search for peer-reviewed studies and datasets linking ion channel blockade to cardiac electrophysiological dysfunctions.
Include preclinical models, clinical data, and computational simulations.
Map Key Event Relationships (KERs):
Collect experimental evidence for causality between successive KEs.
Review dose-response data and time-course relationships to establish quantitative links.
Data Sources:
Databases like PubMed, TOXNET, and the AOP-Wiki platform.
High-throughput screening data and in vitro electrophysiology studies (e.g., patch-clamp assays for hERG).
3. Molecular Initiating Event (MIE) Identification
Pinpoint the blockade of specific cardiac ion channels as the MIE.
Focus on hERG channel inhibition due to its established link with QT prolongation.
Include other ion channels where evidence supports their involvement in arrhythmogenesis.
Assess potential co-factors, such as genetic predispositions (e.g., Long QT Syndrome) or electrolyte imbalances (e.g., hypokalemia).
4. Key Event (KE) Development
Define cellular, tissue, and organ-level KEs that lead to arrhythmia-associated sudden cardiac death.
Examples of KEs:
Prolongation of action potential duration (APD).
Formation of early afterdepolarizations (EADs).
Triggered activity.
Induction of cardiac arrhythmias.
Reduction in cardiac output.
Sudden cardiac death.
Ensure each KE is:
Observable: Measurable in experimental or clinical settings.
Biologically Plausible: Supported by mechanistic evidence.
Relevant: Tied directly to the adverse outcome.
5. Key Event Relationships (KERs)
Develop Evidence Chains: Establish how each KE leads to the next, based on experimental data.
Define Relationship Characteristics:
Biological plausibility.
Empirical support (dose-response, time course, etc.).
Quantitative understanding (e.g., thresholds for APD prolongation leading to EADs).
Address Uncertainties:
Investigate variability across species and individual susceptibility factors.
6. Integration of Quantitative Data
Develop models that link MIEs and KEs quantitatively, where possible.
Use computational tools, such as electrophysiological simulations, to predict arrhythmogenic risk.
Incorporate dose-response and temporal relationship data to build predictive frameworks.
Collaborate with databases like the OECD QSAR Toolbox for integrating chemical-specific data.
Summary of the AOP
Events:
Molecular Initiating Events (MIE)
Key Events (KE)
Adverse Outcomes (AO)
| Type | Event ID | Title | Short name |
|---|
| MIE | 2282 | Cardiac ion channels blockade | Cardiac ion channels (e.g., potassium, sodium, calcium) |
| KE | 1961 | Prolongation of Action Potential Duration | Prolongation of Action Potential |
| KE | 2283 | Increased the early premature depolarizations during repolarization | Early premature depolarizations |
| AO | 1929 | Increased incidence of cardiovascular morbidity and mortality in the general population | Increased incidence of cardiovascular morbidity and mortality |
Relationships Between Two Key Events (Including MIEs and AOs)
| Title | Adjacency | Evidence | Quantitative Understanding |
|---|
| Cardiac ion channels (e.g., potassium, sodium, calcium) leads to Prolongation of Action Potential | adjacent | High | High |
| Prolongation of Action Potential leads to Early premature depolarizations | adjacent | High | Moderate |
| Early premature depolarizations leads to Increased incidence of cardiovascular morbidity and mortality | adjacent | Moderate | Moderate |
Network View
Prototypical Stressors
Life Stage Applicability
| Life stage | Evidence |
|---|---|
| Not Otherwise Specified | Moderate |
Taxonomic Applicability
Sex Applicability
| Sex | Evidence |
|---|---|
| Mixed | High |
Overall Assessment of the AOP
This AOP provides a structured and scientifically plausible framework to understand and predict the cardiotoxicity of compounds. Its assessment is based on biological plausibility, empirical evidence, applicability, and limitations, as summarized below:
Biological Plausibility
The AOP builds on well-established mechanisms of cardiac electrophysiology, where ion channel blockade (e.g., hERG potassium channel inhibition) leads to prolonged action potential duration (APD), early afterdepolarizations (EADs), and arrhythmogenesis.
Strong mechanistic understanding exists for key ion channels (potassium, sodium, and calcium) and their role in maintaining cardiac rhythm.
The causal relationships between key events, such as prolonged APD and EAD formation, are supported by experimental and clinical data.
Empirical Evidence
Robust dose-response relationships between ion channel inhibitors and QT prolongation, a measurable surrogate for APD prolongation.
In vitro studies (e.g., patch-clamp assays) demonstrate a clear link between ion channel inhibition and arrhythmogenic changes in cardiac cells.
In vivo and clinical studies provide evidence for the progression from arrhythmias to sudden cardiac death.
Applicability
The AOP is broadly applicable across drug safety assessment, environmental toxicology, and regulatory frameworks, particularly in screening for cardiotoxic risks of chemicals and pharmaceuticals. It supports the development of non-animal testing methods, such as high-throughput in vitro assays and computational models.
Utility
The AOP facilitates the identification of ion channel blocking liabilities early in drug discovery and development.
It can guide regulatory decision-making, aligning with international safety guidelines (e.g., ICH S7B and E14 for QT prolongation and cardiac safety).
2. Limitations and Uncertainties
Variability in Response
Individual susceptibility to arrhythmias varies due to factors such as genetic predispositions (e.g., Long QT Syndrome), electrolyte imbalances (e.g., hypokalemia), and comorbidities.
Species-specific differences in cardiac electrophysiology may complicate extrapolation of results from animal models to humans.
Data Gaps
Limited quantitative understanding of some Key Event Relationships (KERs), such as the thresholds for EAD formation leading to arrhythmias.
Insufficient data for certain compounds, particularly environmental toxicants, to establish their arrhythmogenic potential.
Complexity of the Outcome
Arrhythmias are influenced by multiple factors, including heart rate, autonomic nervous system regulation, and structural heart diseases, which are not fully captured in the AOP.
The transition from arrhythmias to sudden cardiac death can involve stochastic events, making prediction challenging.
Lack of High-Throughput Tools for Downstream KEs
While ion channel blockade and APD prolongation are measurable using in vitro assays, downstream events like arrhythmias and reduced cardiac output require more complex models.
Domain of Applicability
1. Biological Domain
Taxonomic Applicability:
Primarily applicable to mammalian species, with a strong focus on humans due to the clinical relevance of cardiac arrhythmias and sudden cardiac death.
Experimental data supporting this AOP are predominantly derived from human cardiac cells (e.g., induced pluripotent stem cell-derived cardiomyocytes), animal models (e.g., rodents, rabbits, and dogs), and non-human primates.
Cross-species differences in cardiac ion channel expression and function should be considered, especially when extrapolating results from animal studies to humans.
Life Stage:
Relevant across all life stages but particularly critical for adults and the elderly, who are more susceptible to arrhythmias due to age-related cardiac changes or co-morbidities.
Can also apply to pediatric populations with congenital heart defects or genetic predispositions (e.g., Long QT Syndrome).
Sex:
No sex-specific differences in the core mechanism of ion channel blockade. However, evidence suggests that females may be at a higher risk for drug-induced arrhythmias due to differences in cardiac repolarization and hormonal influences.
2. Chemical Domain
Types of Chemicals:
Pharmaceuticals:
Drugs that block cardiac ion channels, such as hERG channel blockers (e.g., cisapride, dofetilide, terfenadine).
Anti-arrhythmic drugs with off-target effects, including sodium and calcium channel blockers.
Environmental Toxicants:
Chemicals with cardiotoxic potential, such as heavy metals (e.g., arsenic, lead) or industrial pollutants.
Natural Toxins:
Naturally occurring substances, such as marine toxins (e.g., ciguatoxins) that affect ion channels.
New Chemicals:
Applicable in the assessment of novel compounds where ion channel-blocking activity is suspected.
Chemical Properties:
Compounds with physicochemical properties allowing interaction with ion channel binding sites.
Lipophilic drugs are particularly prone to off-target effects on cardiac ion channels.
3. Regulatory Domain
Pharmaceutical Development:
Integral to preclinical safety assessment in the drug discovery pipeline, focusing on ion channel interaction (e.g., ICH S7B guidelines for non-clinical evaluation of QT prolongation).
Supports clinical risk management strategies for arrhythmia-prone drugs (e.g., ICH E14 guidelines for assessing QT interval prolongation in humans).
Environmental Toxicology:
Relevant for evaluating environmental pollutants or industrial chemicals with potential cardiotoxicity.
Can be used in ecological risk assessment to study impacts on non-human species with similar cardiac electrophysiological properties.
Chemical Risk Assessment:
Applicable in regulatory frameworks for the evaluation of consumer products, food additives, and industrial chemicals with cardiotoxic potential.
4. Mechanistic Domain
Ion Channels:
Particularly relevant to compounds that affect:
hERG potassium channels (KCNH2): Most commonly implicated in QT prolongation.
Voltage-gated sodium channels (Nav1.5): Critical for action potential initiation and propagation.
Voltage-gated calcium channels (Cav1.2): Essential for plateau phase and excitation-contraction coupling.
Physiological Processes:
Focused on pathways influencing cardiac electrophysiology, including action potential generation, repolarization, and propagation.
Essentiality of the Key Events
1. Molecular Initiating Event (MIE): Cardiac Ion Channel Blockade
Essentiality: High
The blockade of key cardiac ion channels, particularly hERG potassium channels, initiates the cascade of events in this AOP.
Experimental evidence demonstrates that blocking hERG channels prolongs the action potential duration (APD) and predisposes cardiomyocytes to arrhythmias.
Modulating the MIE (e.g., preventing ion channel blockade through selective drug design) directly prevents downstream events.
2. KE1: Prolongation of Action Potential Duration (APD)
Essentiality: High
Prolonged APD is a hallmark of disrupted cardiac electrophysiology, creating conditions for early afterdepolarizations (EADs).
Pharmacological studies confirm that compounds causing QT interval prolongation (a surrogate marker of prolonged APD) increase the risk of arrhythmias.
Reversing APD prolongation (e.g., with selective ion channel activators) prevents the formation of EADs, highlighting its critical role in the pathway.
3. KE2: Early Afterdepolarizations (EADs)
Essentiality: High
EADs are a direct consequence of prolonged APD and serve as a primary trigger for arrhythmogenic activity.
Experimental studies demonstrate that preventing EADs, either by stabilizing the action potential plateau or enhancing repolarization, interrupts the progression to triggered activity and arrhythmias.
Compounds that block EAD formation (e.g., late sodium current inhibitors) effectively reduce arrhythmia risk.
4. AO: Increased incidence of cardiovascular morbidity and mortality
Essentiality: High
Cardiac arrhythmias, particularly Torsades de Pointes (TdP) or ventricular fibrillation, are the immediate precursors to reduced cardiac output and sudden cardiac death.
Clinical and experimental evidence demonstrates that preventing arrhythmias, either through anti-arrhythmic drugs or defibrillation, directly averts downstream adverse outcomes.
The link between arrhythmias and reduced cardiac output is well-established, reinforcing this KE's critical role.
Evidence Assessment
MIE: Cardiac Ion Channel Blockade
Numerous in vitro studies using patch-clamp assays show that drugs blocking hERG channels cause QT prolongation.
High-throughput screening assays validate the MIE for predicting cardiotoxicity.
Strength: Strong evidence.
KE1: Prolongation of APD
Studies in isolated cardiomyocytes demonstrate that ion channel blockers prolong APD.
Correlation between QT interval prolongation (a surrogate marker) and APD is well-documented in preclinical and clinical settings.
Strength: Strong evidence.
KE2: Early Afterdepolarizations (EADs)
EAD formation is observed in experimental models of prolonged APD, particularly under conditions like hypokalemia or bradycardia.
Pharmacological inhibitors reducing EAD formation prevent arrhythmias, confirming causality.
Strength: Moderate to strong evidence.
AO: Increased incidence of cardiovascular morbidity and mortality (arrhythmias).
Arrhythmias are experimentally and clinically shown to impair cardiac output.
Evidence is indirect, as reduced cardiac output is a downstream consequence of arrhythmias.
Strength: Moderate evidence.
Evidence for Key Event Relationships (KERs)
MIE → KE1 (Cardiac Ion Channel Blockade → Prolongation of APD)
Strong empirical support from in vitro and in vivo studies demonstrating that ion channel blockers prolong APD.
Strength: Strong evidence.
KE1 → KE2 (Prolongation of APD → EAD Formation)
Clear dose-response relationships between prolonged APD and EAD formation in electrophysiological studies.
Strength: Moderate to strong evidence.
KE2 → AO (EAD Formation → Cardiac Arrhythmias)
Triggered activity consistently follows EADs in experimental models, especially under conditions enhancing cellular excitability.
Strength: Moderate evidence.
AO (Triggered Activity → Cardiac Arrhythmias)
Experimental studies and clinical cases show that triggered activity initiates arrhythmias.
Strength: Moderate to strong evidence.
In Vitro Models:
hERG channel inhibition assays (e.g., patch-clamp studies).
Human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and Cardiac Organoid.
In Vivo Models:
Animal models (e.g., guinea pigs, rabbits, or dogs) are used to assess QT prolongation and arrhythmias.
Computational Models:
QSAR models predict the likelihood of ion channel interactions based on the molecular structure of stressors.
Known Modulating Factors
| Modulating Factor (MF) | Influence or Outcome | KER(s) involved |
|---|---|---|
|
Genetic mutations (e.g., LQTS) Electrolyte imbalances Heart rate (bradycardia) Hormonal fluctuations Environmental stressors |
Increased sensitivity to ion channel blockade and arrhythmias Amplifies or mitigates APD prolongation, EADs, and triggered activity Prolongs diastolic intervals, increasing EAD risk Modulate ion channel activity and arrhythmogenic potential Alter cardiac electrophysiology (e.g., hypoxia increases arrhythmogenic risk) |
MIE → KE1, KE1 → KE2 KE1 → KE2 KE1 → KE2 KE2 → AO |
Quantitative Understanding
- Dose-response relationships are well-established for MIE → KE1 (ion channel blockade → APD prolongation).
- Temporal concordance between KE1 → KE2 (APD prolongation → EAD formation) is supported by in vitro studies or Cardiac organoid.
- Clinical QT prolongation thresholds provide a quantitative basis for predicting cadiac toxicity risk.
Considerations for Potential Applications of the AOP (optional)
This Adverse Outcome Pathway (AOP) provides a mechanistic framework for understanding and predicting the cardiotoxic effects of chemicals and pharmaceuticals. The AOP outlines a sequence of key events (KEs), starting with the molecular initiating event (MIE) of cardiac ion channel blockade, which leads to prolonged action potential duration (APD), early afterdepolarizations (EADs), triggered activity, cardiac arrhythmias, and ultimately, sudden cardiac death. Strong biological plausibility, supported by empirical evidence, underpins the relationships between these events, although quantitative understanding remains moderate for certain transitions. The domain of applicability spans mammalian species, particularly humans, with broad relevance across life stages and both sexes. Modulating factors, including genetic predispositions (e.g., Long QT Syndrome), electrolyte imbalances, co-administration of drugs, and environmental stressors, significantly influence the progression of KEs and the likelihood of the adverse outcome. The AOP is well-suited for regulatory applications, such as drug safety assessment, chemical risk evaluation, and integration into frameworks like the OECD Integrated Approaches to Testing and Assessment (IATA). It also supports research applications, including mechanistic toxicology, the development of alternative testing methods, and personalized medicine. Despite its strengths, limitations such as inter-individual variability, species differences, and data gaps for certain chemical classes highlight the need for further refinement. By addressing these challenges, the AOP can enable more predictive risk assessments, guide regulatory decision-making, and facilitate the safe development of drugs and industrial chemicals. This comprehensive framework underscores the value of AOPs in advancing public health and improving safety evaluation processes.
References
Sanguinetti, M. C., & Tristani-Firouzi, M. (2006). hERG potassium channels and cardiac arrhythmia. Nature, 440(7083), 463–469.
Roden, D. M. (2004). Drug-induced prolongation of the QT interval. New England Journal of Medicine, 350(10), 1013–1022.
Curran, M. E., et al. (1995). A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell, 80(5), 795–803.
Vandenberg, J. I., et al. (2012). hERG K+ channels: Structure, function, and role in cardiac arrhythmia. Physiological Reviews, 92(3), 1393–1478.
Kannankeril, P., Roden, D. M., & Darbar, D. (2010). Drug-induced long QT syndrome. Pharmacological Reviews, 62(4), 760–781.
Clancy, C. E., & Rudy, Y. (1999). Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature, 400(6744), 566–569.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2005). ICH S7B: Nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2005). ICH E14: Clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs.
Villeneuve, D. L., et al. (2014). Adverse outcome pathway (AOP) development I: Strategies and principles. Toxicological Sciences, 142(2), 312–320.
OECD. (2018). Users' Handbook Supplement to the Guidance Document for Developing and Assessing Adverse Outcome Pathways. OECD Series on Adverse Outcome Pathways, No. 1. Paris: OECD Publishing.
Cheng, H., et al. (2012). Cardiomyocytes derived from human pluripotent stem cells: A model for studying the cardiac channelopathy long-QT syndrome. Science Translational Medicine, 4(130), 130ra49.
Milnes, J. T., et al. (2010). hERG K+ channel pharmacology: Insights into mechanisms of drug-induced QT prolongation and arrhythmia. Journal of Pharmacological and Toxicological Methods, 61(3), 171–180.
Zipes, D. P., & Wellens, H. J. (1998). Sudden cardiac death. Circulation, 98(21), 2334–2351.
Di Diego, J. M., & Antzelevitch, C. (2009). Cellular basis for the electrocardiographic manifestations of the long QT syndrome: Insights from modeling and experimental studies. Circulation Research, 105(7), 707–721.
Schwartz, P. J., et al. (2013). Clinical aspects of inherited long-QT syndrome: A changing paradigm. Journal of the American College of Cardiology, 62(20), 2017–2022.
Tester, D. J., & Ackerman, M. J. (2007). Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. Journal of the American College of Cardiology, 49(2), 240–246.
O’Hara, T., Virág, L., & Rudy, Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Computational Biology, 7(5), e1002061.
Mirams, G. R., et al. (2011). Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovascular Research, 91(1), 53–61.
Roden, D. M. (2008). Cellular basis of drug-induced torsades de pointes: From ion channel to clinical disorder. Cardiovascular Research, 68(1), 137–146.
Moss, A. J., & Kass, R. S. (2005). Long QT syndrome: From channels to cardiac arrhythmias. Journal of Clinical Investigation, 115(8), 2018–2024.
Ankley, G. T., et al. (2010). Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environmental Toxicology and Chemistry, 29(3), 730–741.
Perkins, E. J., et al. (2015). Adverse outcome pathways for regulatory applications: Examining North American and European experience. Environmental Toxicology and Chemistry, 34(9), 1957–1973.