
This AOP is licensed under the BY-SA license. This license allows reusers to distribute, remix, adapt, and build upon the material in any medium or format, so long as attribution is given to the creator. The license allows for commercial use. If you remix, adapt, or build upon the material, you must license the modified material under identical terms.
AOP: 472
Title
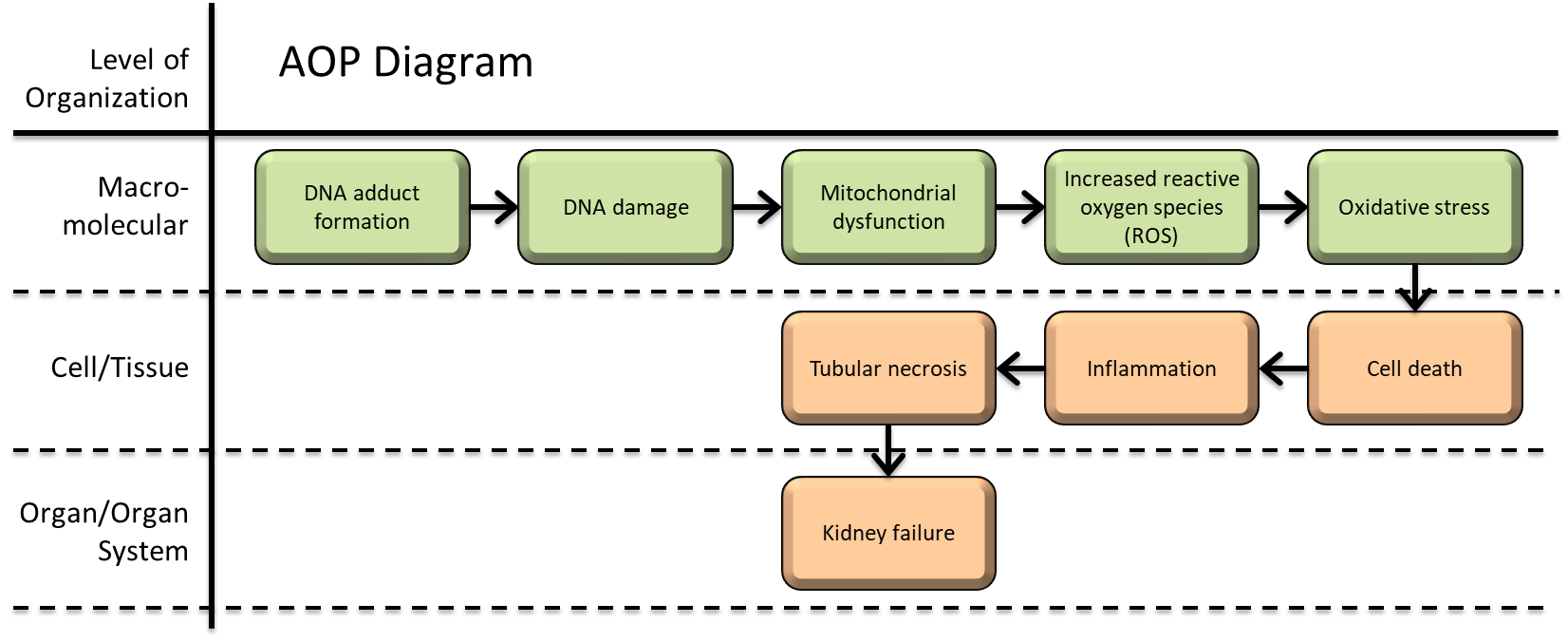
DNA adduct formation leading to kidney failure
Short name
Graphical Representation
Additional AOP Exploration Options
Click links below to explore AOP 472, DNA adduct formation leading to kidney failure in tools offered by third parties.
Point of Contact
Contributors
- Manoe Janssen
- Devon Barnes
- Huan Yang
Coaches
OECD Information Table
| OECD Project # | OECD Status | Reviewer's Reports | Journal-format Article | OECD iLibrary Published Version |
|---|---|---|---|---|
This AOP was last modified on March 29, 2026 04:26
Revision dates for related pages
| Page | Revision Date/Time |
|---|---|
| Formation, Pro-mutagenic DNA Adducts | September 16, 2017 10:15 |
| Increase, DNA damage | May 08, 2019 12:28 |
| Increase, Mitochondrial dysfunction | February 11, 2026 07:06 |
| Increase, Reactive oxygen species | June 12, 2025 01:27 |
| Increase, Oxidative Stress | February 11, 2026 07:05 |
| Increase, Cell death | November 27, 2024 11:26 |
| Increase, Inflammation | February 28, 2024 06:33 |
| Occurrence, renal proximal tubular necrosis | September 16, 2017 10:16 |
| Increased, Kidney Failure | January 16, 2019 08:57 |
| Formation, Pro-mutagenic DNA Adducts leads to Increase, DNA Damage | March 29, 2026 04:17 |
| Increase, DNA Damage leads to Increase, Mitochondrial dysfunction | March 29, 2026 04:17 |
| Increase, Mitochondrial dysfunction leads to Increase, ROS | April 11, 2024 15:22 |
| Increase, ROS leads to Increase, Oxidative Stress | June 23, 2026 06:57 |
| Increase, Oxidative Stress leads to Increase, Inflammation | October 19, 2023 09:39 |
| Increase, Inflammation leads to Increase, Cell death | March 29, 2026 04:21 |
| Increase, Cell death leads to Occurrence, renal proximal tubular necrosis | March 29, 2026 04:24 |
| Occurrence, renal proximal tubular necrosis leads to Increased, Kidney Failure | March 29, 2026 04:25 |
| Cisplatin | February 03, 2022 11:34 |
| Oxaliplatin | March 29, 2026 04:27 |
| Carboplatin | March 29, 2026 04:27 |
Abstract
This Adverse Outcome Pathway (AOP) depicts a possible sequence of key events that associate how DNA adduct formation caused by platinum anticancer drugs can induce tubular necrosis, resulting in the occurrence of kidney failure as the adverse outcome (AO). Currently, cisplatin, carboplatin and oxaliplatin are the three most utilised Pt-based drugs used globally for the treatment of cancer. The cytotoxicity of these agents are primarily determined by their DNA adducts. Increased intracellular concentrations following uptake of platinum anticancer agents into the nephron, mostly by proximal convoluted tubules, evoking a cascade of negative cellular responses that can cause tubular necrosis, potentially resulting in kidney failure. The aquation of platinum anticancer agents following cellular uptake produces electrophilic intermediates that covalently bind to nucleophilic sites on DNA to form adducts that represent the molecular initiating event (MIE). The nephrotoxic response following the formation of DNA adducts leads to DNA damage (KE1) and mitochondrial dysfunction (KE2). These events promote the release of reaction oxygen species (ROS) (KE3) to induce oxidative stress (KE4), causing cell death (KE5) and inflammation (KE6). As these cells detach from the basement membrane, they are deposited in the tubular lumen. Tubular obstruction and inflammatory responses to proximal tubule insult can cause secondary toxicity and tubular necrosis (KE7), further amplifying kidney injury and a progressive decline of function, finally resulting in kidney failure (AO). This information is primarily based on mechanisms of actions previously described in cited literature sources and intended as a resource template for AOP development and data organization. The primary species of this AOP is humans, most often observed following chemotherapeutic intervention when treating a range of tumours and multiple malignancies; supported by data with potential applicability for other species.
AOP Development Strategy
Context
DNA adduct formation leading to kidney failure
This Adverse Outcome Pathway (AOP) details the sequence of key events (KE) that connect DNA adduct formation to kidney failure. Platinum (Pt)-based antineoplastics that are routinely utilized as anticancer agents for the effective treatment of multiple cancer types, including breast, cervical, oesophageal, bladder, small cell lung and testicular cancer (1), can function as chemical stressors for the described pathway. Currently, cisplatin, carboplatin and oxaliplatin are the three most utilised Pt-based drugs used globally for the treatment of cancer, all possessing a similar chemical structure consisting of platinum, carrier and leaving groups, undergoing similar mechanisms of cytotoxicity. Renowned for being one of the most potent and effective chemotherapeutics available, preventing cancerous cells from multiplying by binding DNA strands, clinical application of Pt anticancer agents remains limited due to the moderate to life-threating severity of their adverse side effects, such as neurotoxicity, hepatoxicity, ototoxicity, cardiotoxicity and myelosuppression and dose-limiting nephrotoxicity (1, 2).
Nephrotoxicity associated with platinum antineoplastic chemotherapeutic intervention
Within the kidney, the proximal tubular cells are most sensitive to the toxic effects of Pt-based drugs. They are responsible for the reabsorption of most nutrients and low molecular weight proteins, and approximately 70% of filtered solutes and water. Furthermore, they use active transport to clear the blood of toxic side products and drug compounds. Uptake of these compounds takes place at the basolateral side of the cells which is exposed to the blood. After entering the cells compounds are again secreted at the apical side of the cells into the kidney filtrate and leave the body with the urine. The toxicity of xenobiotics is directly related to their exposure to proximal tubule cells, their uptake through transport proteins like the SLC22A2/organic cation transporter 2 (OCT2), there reactivity in the cytoplasm and secretion through efflux transporters like multi-antimicrobial extrusion protein transporter-1 (MATE1) and multidrug resistance-associated protein 2 (MRP2) (3). This also implies that (genetic) variations in drug transporter activity can have implications in an individual’s susceptibility to (Pt)-based antineoplastics (4). Over time, several generations of Pt-based antineoplastics have been developed to make this group of compounds less nephrotoxic. Cisplatin was the first generation of Pt-based antineoplastics approved by the U.S. Food and Drug administration (FDA) in 1978. Classified as an alkylating agent, cisplatin was shown to inhibit cell division of bacteria and was developed as a treatment for mesotheliomas (5). However, issues arose following reports of cisplatin-induced nephrotoxicity, most notably the prevalence of acute kidney injury (AKI) and acute tubular necrosis (ATN) following treatment (6, 7). This increased susceptibility to severe nephrotoxicity during these early clinical trials led to the revision of cisplatin as an applicable chemotherapeutic agent. Therefore, much of the initial attempts of Pt drug development was to determine less-toxic analogues of cisplatin that retained similar levels of anticancer activity. The second-generation Pt drug, carboplatin, was developed to decrease the dose-limiting toxicity of cisplatin. Carboplatin’s mechanism of action is similar to that of cisplatin, however it was shown to have fewer and less severe side effects (8), benefiting from a reduced aquation rate due to its bidentate cyclobutane dicarboxylate ligand (9), and later approved by the FDA for use in 1989. Due to its decreased nephrotoxicity, carboplatin was identified as a more suitable option for aggressive, high-dose chemotherapy. However, carboplatin was also dose-limited following reports of cumulative anaemia and myelosuppression (10). Carboplatin is also a nephrotoxic drug, albeit much lower compared to cisplatin due to its increased stability (11). Nephrotoxicity had been reported in patients treated with intraperitoneal carboplatin, high-dose carboplatin, or in combination with other drugs (12), with AKI also shown to occur within days and often only partially reversible (13). The third-generation Pt drug oxaliplatin was developed to overcome both cisplatin and carboplatin toxicity. Oxaliplatin possesses a Pt complex with (1R,2R)-1,2-diaminocyclohexane (DACH) ligand and oxalate functioning as a leaving group. The bidentate oxalate considerably decreases the reactivity of oxaliplatin, limiting toxicity (14). However, although less nephrotoxic than both cisplatin and carboplatin, oxaliplatin can still induce proximal tubular cell damage (15). Furthermore, oxaliplatin has also been reported to cause several forms of nephrotoxicity, such as kidney tubular vacuolization (16) and acidosis (17-19). Despite the reduced incidence of nephrotoxicity, carboplatin and oxaliplatin, along with cisplatin, have all been reported to induce AKI and thrombotic microangiopathy (20).
References
1. Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 2018;47(19):6645-53.
2. Markman M. Toxicities of the platinum antineoplastic agents. Expert Opin Drug Saf. 2003;2(6):597-607.
3. McSweeney KR, Gadanec LK, Qaradakhi T, Ali BA, Zulli A, Apostolopoulos V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers (Basel). 2021;13(7).
4. Zazuli Z, Vijverberg S, Slob E, Liu G, Carleton B, Veltman J, et al. Genetic Variations and Cisplatin Nephrotoxicity: A Systematic Review. Front Pharmacol. 2018;9:1111.
5. Brown A, Kumar S, Tchounwou PB. Cisplatin-Based Chemotherapy of Human Cancers. J Cancer Sci Ther. 2019;11(4).
6. DeConti RC, Toftness BR, Lange RC, Creasey WA. Clinical and pharmacological studies with cis-diamminedichloroplatinum (II). Cancer Res. 1973;33(6):1310-5.
7. Eustace P. History and development of cisplatin in the management of malignant disease. Cancer Nurs. 1980;3(5):373-8.
8. Hydes PC, Russell MJ. Advances in platinum cancer chemotherapy. Advances in the design of cisplatin analogues. Cancer Metastasis Rev. 1988;7(1):67-89.
9. Calvert AH, Harland SJ, Newell DR, Siddik ZH, Jones AC, McElwain TJ, et al. Early clinical studies with cis-diammine-1,1-cyclobutane dicarboxylate platinum II. Cancer Chemother Pharmacol. 1982;9(3):140-7.
10. Suzuki K, Matsumoto K, Hashimoto K, Kurokawa K, Jinbo S, Suzuki T, et al. Carboplatin-based combination chemotherapy for testicular cancer: relationship among administration dose of carboplatin, renal function and myelosuppression. Hinyokika Kiyo. 1995;41(10):775-80.
11. Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63(1):12-31.
12. English MW, Skinner R, Pearson AD, Price L, Wyllie R, Craft AW. Dose-related nephrotoxicity of carboplatin in children. Br J Cancer. 1999;81(2):336-41.
13. Deray G, Ben-Othman T, Brillet G, Baumelou B, Gabarre J, Baumelou A, et al. Carboplatin-induced acute renal failure. Am J Nephrol. 1990;10(5):431-2.
14. Kidani Y, Inagaki K, Iigo M, Hoshi A, Kuretani K. Antitumor activity of 1,2-diaminocyclohexane--platinum complexes against sarcoma-180 ascites form. J Med Chem. 1978;21(12):1315-8.
15. Haschke M, Vitins T, Lude S, Todesco L, Novakova K, Herrmann R, et al. Urinary excretion of carnitine as a marker of proximal tubular damage associated with platin-based antineoplastic drugs. Nephrol Dial Transplant. 2010;25(2):426-33.
16. Joybari AY, Sarbaz S, Azadeh P, Mirafsharieh SA, Rahbari A, Farasatinasab M, et al. Oxaliplatin-induced renal tubular vacuolization. Ann Pharmacother. 2014;48(6):796-800.
17. Sonnenblick A, Meirovitz A. Renal tubular acidosis secondary to capecitabine, oxaliplatin, and cetuximab treatment in a patient with metastatic colon carcinoma: a case report and review of the literature. Int J Clin Oncol. 2010;15(4):420-2.
18. Negro A, Grasselli C, Galli P. Oxaliplatin-induced proximal renal tubular acidosis. Intern Emerg Med. 2010;5(3):267-8.
19. Linch M, Cunningham D, Mingo O, Stiles A, Farquhar-Smith WP. Renal tubular acidosis due to oxaliplatin. Ann Oncol. 2007;18(4):805-6.
20. Gupta S, Portales-Castillo I, Daher A, Kitchlu A. Conventional Chemotherapy Nephrotoxicity. Adv Chronic Kidney Dis. 2021;28(5):402-14 e1.
Strategy
This AOP is being developed as part of the ONTOX consortium. The aim of this consortium is to provide a generic strategy to create innovative new approach methodologies (NAMs) in order to predict systemic repeated dose toxicity effects that, upon combination with tailored exposure assessment, will enable human risk assessment. Part of this approach is the development of physiological maps, quantitative adverse outcome pathway networks and ontology frameworks. This project is funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement No 963845. https://ontox-project.eu/project/
Within the ONTOX consortium in vitro test batteries are being developed to evaluate toxicity in the liver, (steatosis and cholestasis), kidneys (tubular necrosis and crystallopathy) and developing brain (neural tube closure and cognitive function defects). This AOP focusses on kidney tubular necrosis as a result of exposure to a DNA-adduct forming compound (Pt-based drugs).
Summary of the AOP
Events:
Molecular Initiating Events (MIE)
Key Events (KE)
Adverse Outcomes (AO)
| Type | Event ID | Title | Short name |
|---|
| MIE | 373 | Formation, Pro-mutagenic DNA Adducts | Formation, Pro-mutagenic DNA Adducts |
| KE | 1194 | Increase, DNA damage | Increase, DNA Damage |
| KE | 177 | Increase, Mitochondrial dysfunction | Increase, Mitochondrial dysfunction |
| KE | 1115 | Increase, Reactive oxygen species | Increase, ROS |
| KE | 1392 | Increase, Oxidative Stress | Increase, Oxidative Stress |
| KE | 1825 | Increase, Cell death | Increase, Cell death |
| KE | 149 | Increase, Inflammation | Increase, Inflammation |
| KE | 1097 | Occurrence, renal proximal tubular necrosis | Occurrence, renal proximal tubular necrosis |
| AO | 759 | Increased, Kidney Failure | Increased, Kidney Failure |
Relationships Between Two Key Events (Including MIEs and AOs)
| Title | Adjacency | Evidence | Quantitative Understanding |
|---|
| Formation, Pro-mutagenic DNA Adducts leads to Increase, DNA Damage | adjacent | High | High |
| Increase, DNA Damage leads to Increase, Mitochondrial dysfunction | adjacent | High | High |
| Increase, Mitochondrial dysfunction leads to Increase, ROS | adjacent | Moderate | Moderate |
| Increase, ROS leads to Increase, Oxidative Stress | adjacent | High | High |
| Increase, Oxidative Stress leads to Increase, Inflammation | adjacent | Moderate | Moderate |
| Increase, Inflammation leads to Increase, Cell death | adjacent | Moderate | Moderate |
| Increase, Cell death leads to Occurrence, renal proximal tubular necrosis | adjacent | High | High |
| Occurrence, renal proximal tubular necrosis leads to Increased, Kidney Failure | adjacent | Moderate | Moderate |
Network View
Prototypical Stressors
Life Stage Applicability
Taxonomic Applicability
Sex Applicability
Overall Assessment of the AOP
Domain of Applicability
Mechanistic evidence for KEs and KERs in this AOP are predominantly sourced from rodent studies, namely mouse and rat, however, it has not yet been reported whether there are limits to the extractability of this AOP to other species and/or life stages. The described AOP presents an overview of the general mechanisms that lead to kidney failure in a range of species, including human, rat, mouse, and hamster.
This AOP can theoretically be applied to all-life stages to all organisms that are able to experience DNA-damage induced kidney failure, unless otherwise stated. There is a high amount of empirical support, with large swathes of studies available detailing the KEs and KERs, however there is limited information regarding the dose- and time-dependent response relationships of some of the stressors for this AOP. This is especially true concerning studies that investigate the measurable effects of the stressors in single studies. This AOP will require additional quantitative data before it can be considered for regulatory significance.
Essentiality of the Key Events
There are many studies using pharmacological inhibitors, dominant-negative mutations, and genetic knockouts, along with blocking OCT2-mediated uptake, that demonstrate how obstructing upstream effectors of Pt drug-induced nephrotoxicity limits kidney injury. For example, limiting the cell death signalling that occurs following Pt drug administration via p53 activation can reduce apoptosis following DNA damage. P53-deficient mice showed clear resistance to cisplatin-induced kidney injury, showing only small increases to BUN and sCr following administration (1). Similarly, an in vitro study using primary renal proximal tubule epithelial cells (RPTECs) demonstrated that cisplatin-induced apoptosis is mediated by p53 and caspase 3 activation independent of either caspase 8, 9 or mitochondrial dysfunction (2). Pifithrin-α, a pharmacological inhibitor of p53, was used to suppress cisplatin-induced apoptosis in a dose-dependent manner, highlighting the role of early p53 phosphorylation and induction in cisplatin-induced cell death (3). Studies have shown that blocking the cell death pathways can alleviate Pt-drug-induced nephrotoxicity and kidney cell death. Knockout of both Fas and TNFR receptors in mice was shown to reduce apoptosis following cisplatin administration (4), whereas apoptotic and necrotic cell death was also reduced in TNFR2 deficient mice (5). Similarly, BUN and sCr levels could be preserved significantly in Bax-knockout mice (6). A decrease in apoptotic cells, tissue damage and cytochrome c release were also observed in these Bax-deficient mice. These findings were then replicated in vitro using primary cultures of proximal tubular cells prepared from these animals. Nutlin-3, an MDM2 antagonist, was also able to suppress mitochondrial events of apoptosis following cisplatin treatment, including Bax activation and cytochrome c release (7). Numerous studies have shown that antioxidants can be used to provide renoprotection by limiting ROS generation negative effects induced by oxidative stress. For example, reducing GSH levels has been shown to be effective at protecting against Pt-drug induced nephrotoxicity (8). The delivery of antioxidants that target the mitochondria can be used to prevent Pt-drug nephrotoxicity by decreasing toxic oxidative stress injury. A combinatory treatment using ebselen and allopurinol, a xanthine oxidase inhibitor, significantly diminished ROS generation, preventing cisplatin-induced nephrotoxicity in rats (9). Similarly, dimethylthiourea, a hydroxyl radical scavenger, prevented increases to BUN and sCr levels and reduced apoptosis, whilst maintaining ATP content, electrochemical potential, NAPDH and GSH levels following cisplatin administration (10). Inhibition of the inflammatory response by IL-10 and suppression of TNFα have been shown to protect the kidneys when administering Pt drugs. Blocking TNFα activity prevented the upregulation of a multitude of additional cytokines all shown to contribute to Pt drug nephrotoxicity (11). Salicylate, an anti-inflammatory agent, was also shown to ameliorate Pt drug nephrotoxicity via its inhibition of TNFα production (12). Together, these studies suggest that Pt-drug nephrotoxicity and kidney cell death can be ameliorated by targeting a multitude of pharmacologic, molecular, and genetic targets.
1. Wei Q, Dong G, Yang T, Megyesi J, Price PM, Dong Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol. 2007;293(4):F1282-91.
2. Cummings BS, Schnellmann RG. Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther. 2002;302(1):8-17.
3. Jiang M, Yi X, Hsu S, Wang CY, Dong Z. Role of p53 in cisplatin-induced tubular cell apoptosis: dependence on p53 transcriptional activity. Am J Physiol Renal Physiol. 2004;287(6):F1140-7.
4. Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, et al. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003;63(1):72-82.
5. Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol. 2003;285(4):F610-8.
6. Wei Q, Dong G, Franklin J, Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007;72(1):53-62.
7. Jiang M, Pabla N, Murphy RF, Yang T, Yin XM, Degenhardt K, et al. Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J Biol Chem. 2007;282(4):2636-45.
8. Zunino F, Pratesi G, Micheloni A, Cavalletti E, Sala F, Tofanetti O. Protective effect of reduced glutathione against cisplatin-induced renal and systemic toxicity and its influence on the therapeutic activity of the antitumor drug. Chem Biol Interact. 1989;70(1-2):89-101.
9. Lynch ED, Gu R, Pierce C, Kil J. Reduction of acute cisplatin ototoxicity and nephrotoxicity in rats by oral administration of allopurinol and ebselen. Hear Res. 2005;201(1-2):81-9.
10. Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim Biophys Acta. 2006;1762(2):256-65.
11. Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110(6):835-42.
12. Ramesh G, Reeves WB. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-alpha. Kidney Int. 2004;65(2):490-9.
Evidence Assessment
Scientific evidence linking nephrotoxicity caused by Pt anticancer agents with kidney failure is robust and dependable. However, the MIE is not specific for nephrotoxicity as it has also been well documented for a range of adverse effects in other pathologies. The site of toxicity can also be determined by the kinetics of the individual stressors, often determined by the respective transporters they use to enter the cell and the number of mitochondria present. There are presently no reported alternative mechanisms that deviate from the postulated AOP, however additional contributors to off-target effects within the AOP should not be excluded.
This AOP is only a qualitative description of the pathway and does not presently include quantitative information on dose-response relationships. There are studies available for each of the KEs and KERs to provide empirical support for this AOP, however there remains a lack of substantial information on the dose-response relationships for some of the individual stressors. As such there is also no singular study that measures all reported KEs simultaneously following exposure to a variety of doses, impeding the capacity to perform a highly accurate dose-response and concordance analysis. Further research will be required to provide an improved quantitative understanding of dose-response relationships between upstream and downstream KEs.
Known Modulating Factors
| Modulating Factor (MF) | Influence or Outcome | KER(s) involved |
|---|---|---|
Quantitative Understanding
Weight of evidence (WoE) analysis indicates that many KERs still lack conclusive experimental evidence, especially toward indicating biological plausibility definitively linking downstream to upstream KEs within individual studies. However, analysis of experimental data supports the overall AOP framework. This AOP should remain considered as a qualitative contribution until quantitative data can be ascertained toward investigating dose-response relationships.
Some quantitative connections have been detailed between upstream KEs, however the variation of both the testing models and posology utilized restricts the possibility to draw comparisons between studies. Due to this, it is evidently more obstinate to assess quantitative relationships between later downstream KEs. Optimally, future efforts to strengthen WoE should aim to rely on data from a singular system type that demonstrates exposure to a stressor that would correlate with changes observed for all described KEs.
Considerations for Potential Applications of the AOP (optional)
The described AOP aims to deliver a mechanistic framework toward the advancement of in vitro assays that proficiently predict quantitative points of departure toward nephrotoxicity safety assessment. These assays could potentially form part of an integrated testing strategy to reduce the need for repeated dose toxicity studies. Producing quantitative data by measuring all KEs in a singular model following repeated, long-term exposure to a variety of concentrations of these chemical initiators could also expedite the advancement of computational predictive approaches. With prospective overlap between KEs, this AOP could provide further connections for expansive AOP networks toward modelling nephrotoxicity and kidney failure.