AOP ID and Title:
Graphical Representation
Status
| Author status | OECD status | OECD project | SAAOP status |
|---|---|---|---|
| Under development: Not open for comment. Do not cite |
Coaches
- Shihori Tanabe
Abstract
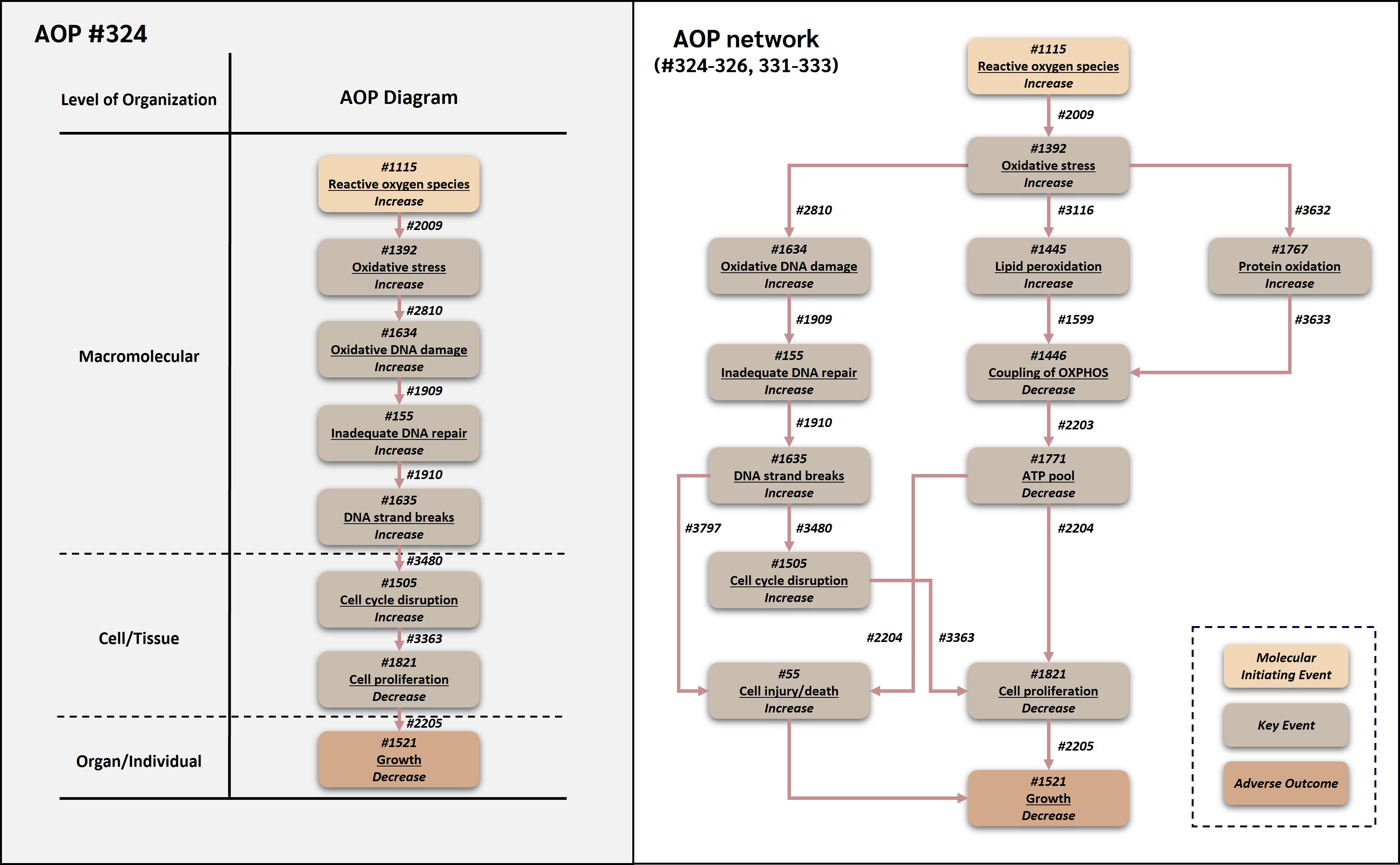
This adverse outcome pathway (AOP 324) describes a linear genotoxic route by which increased reactive oxygen species (ROS) can lead to decreased organismal growth. In this AOP, increased ROS is treated operationally as the molecular initiating event (MIE) because it represents the earliest common measurable redox perturbation shared by many stressors within the broader ROS-mediated AOP networks. Increased ROS leads to oxidative stress, which causes oxidative DNA damage. When oxidative DNA lesions exceed or impair repair capacity, inadequate DNA repair can occur, allowing DNA strand breaks to accumulate. Strand breaks activate cell cycle checkpoint responses and disrupt progression through the cell cycle. Sustained cell cycle disruption reduces cell proliferation, and reduced cell proliferation ultimately contributes to decreased growth.
The AOP reuses and connects existing AOP-Wiki components, including key events (KEs) and key event relationships (KERs) from OECD/WPHA-WNT endorsed AOPs. In particular, the oxidative DNA damage module is derived from AOP 296, the oxidative stress and DNA damage context is supported by AOP 478, the cell-cycle disruption KE is shared with AOP 212, and the link from decreased cell proliferation to decreased growth is reused from AOP 263 (AOP-Wiki, 2026a, 2026b, 2026c, 2026d; OECD, 2022, 2023; Carrothers et al., 2025). The AOP is biologically plausible across aerobic eukaryotes because ROS metabolism, antioxidant defenses, DNA damage response pathways, DNA repair, cell cycle regulation, and growth processes are broadly conserved. Empirical support is derived from studies in algae, fish embryos and cell lines, mammalian cells, and other systems exposed to oxidative or genotoxic stressors. The AOP is expected to be useful for mechanistic interpretation of oxidative stress-related toxicity, support of integrated approaches to testing and assessment (IATA), and prioritization of stressors that produce oxidative DNA damage and growth impairment.
Acknowledgement
This project was funded by the Research Council of Norway (RCN), grant no. RCN-315929 “EXPECT: In silico and experimental screening platform for characterizing environmental impact of industry development in the Arctic” (https://www.niva.no/en/projects/expect), the European Partnership for the Assessment of Risks from Chemicals (PARC) through European Union’s Horizon Europe research and innovation programme (Grant Agreement No 101057014, and supported by the NIVA Computational Toxicology Program, NCTP (https://www.niva.no/en/featured-pages/nctp, grant. No. RCN-342628).
AI disclosure
Artificial intelligence (AI) tools were used to support literature prioritization, review and AOP-Wiki page preparation in this work. AOP-helpFinder was used for automated literature mining, and ChatGPT (OpenAI) was used as an auxiliary tool for title and abstract screening, extraction of study metadata, and identification of potential weight-of-evidence indicators. AI-assisted outputs were used only to organize and prioritize information and were verified against the original sources by the authors before inclusion. Additional AI assistance was used for formatting, copy-editing, citation cross-checking, and harmonization of the AOP-Wiki pages. All scientific interpretations, weight-of-evidence judgments, final wording, and conclusions were determined and approved by the authors, who take full responsibility for the content and integrity of the work.
AOP Development Strategy
Context
ROS are continuously formed during aerobic metabolism and are also generated in response to environmental stressors. At controlled levels, ROS participate in redox signaling, while excessive ROS can disturb redox homeostasis and initiate oxidative damage to cellular macromolecules (Schieber and Chandel, 2014; Sies et al., 2017). DNA is a major target of oxidative attack. Oxidative DNA lesions such as 8-oxo-2'-deoxyguanosine and other oxidized bases can arise endogenously or following toxic insult, and these lesions may contribute to mutation, strand break formation, and activation of DNA damage responses if they are not repaired correctly or efficiently (Cooke et al., 2003; OECD, 2023).
This AOP was developed to represent the DNA damage-driven linear route within the broader ROS-growth AOP network. The route was selected because oxidative DNA damage is a well-established consequence of oxidative stress and because downstream events such as inadequate DNA repair, strand breaks, cell cycle disruption, and reduced cell proliferation provide a mechanistically coherent bridge between molecular damage and decreased organismal growth (Cuddihy and O'Connell, 2003; Conlon and Raff, 1999; OECD, 2022; OECD, 2023). The AOP was therefore designed to provide a focused, reviewable linear representation of one major mechanistic branch by which excessive ROS can impair growth.
Strategy
AOP 324 was developed using the principles described in OECD AOP guidance, including modular description of KEs and KERs, reuse of existing AOP-Wiki content where appropriate, evidence evaluation using biological plausibility, empirical support, essentiality, and quantitative understanding, and clear description of the biological domain of applicability (OECD, 2018, 2021). The intent was not to create a fully de novo set of biological objects, but to assemble a linear AOP from established and reusable AOP-Wiki components wherever possible. This modular strategy is important because the AOP is part of a broader ROS-growth AOP network and because its individual KEs and KERs are expected to be reused in other oxidative stress, genotoxicity, and growth impairment AOPs.
Existing AOP-Wiki content and OECD-endorsed AOPs were reviewed at an early stage to identify KEs and KERs that could be reused directly, adapted, or used as supporting context. AOP 296 was the primary source for the oxidative DNA damage module. It is an endorsed AOP describing oxidative DNA damage leading to chromosomal aberrations and mutations and includes KE 1634 (Increase, Oxidative DNA damage), KE 155 (Inadequate DNA repair), KE 1635 (Increase, DNA strand breaks), and relationships involving oxidative DNA damage, inadequate DNA repair, and strand break formation (AOP-Wiki, 2026b; OECD, 2023). AOP 478, an endorsed AOP on deposition of energy leading to cataracts, provided additional support for the upstream oxidative stress context, including KE 1392 (Oxidative stress), KE 1634, KE 155, KE 1635, and the relationship from oxidative stress to oxidative DNA damage in a radiation-relevant setting (AOP-Wiki, 2026a; Carrothers et al., 2025). AOP 212 was reviewed because it contains KE 1505 (Cell cycle, disrupted) and evidence that disruption of cell-cycle regulation can lead to downstream changes in cell fate (AOP-Wiki, 2026c). AOP 263 was reviewed because it is an endorsed AOP linking decreased cell proliferation to decreased growth, and AOP 324 reuses the downstream KE 1821 (Decrease, Cell proliferation), AO 1521 (Decrease, Growth), and KER 2205 (Decrease, Cell proliferation leads to Decrease, Growth) from that AOP (AOP-Wiki, 2026d; OECD, 2022; Song and Villeneuve, 2021).
In accordance with the OECD AOP coaching checklist (ver. 2024-10-30), the lead author (Y. Song) have communicated with the authors of the following existing KEs and KERs that are reused or expanded in this AOPN: KE 1115 (Mystery of ROS consortium, coordinated by S. Tanabe, NIHS Japan); KE 1392, KE 1634, KE 155, KE 1635, KER 2810, KER 1909, KER 1910 (AOP 296 and AOP 478: C. Yauk, University of Ottawa; V. Chauhan, Health Canada); KE 1505 (AOP 212: S. Tanabe, NIHS Japan); KE 55 (AOPs 12, 13, 17, 38, 48: relevant corresponding authors notified); KE 1446, KE 1771, KE 1821, KE 1521, KER 2203, KER 2204, KER 2205 (AOP 263: Y. Song, NIVA). Notification documentation is available from the corresponding author on request.
The resulting AOP 324 therefore represents an upstream extension and re-routing of established AOP-Wiki knowledge. Compared with AOP 296, AOP 324 extends upstream from oxidative DNA damage to increased ROS and oxidative stress and extends downstream from DNA damage response biology to decreased cell proliferation and growth. Compared with AOP 263, it reuses the final growth-relevant segment but provides a different upstream causal route, namely oxidative DNA damage and cell-cycle disruption rather than mitochondrial uncoupling and ATP depletion. Compared with AOP 478, it reuses the oxidative stress-DNA damage portion of the radiation AOP but directs that conserved genotoxic sequence toward growth inhibition rather than cataract formation. Compared with AOP 212, it reuses cell-cycle disruption as a modular KE but places it in the context of DNA strand break-mediated checkpoint activation rather than histone deacetylase inhibition.
The literature review and evidence assembly process followed an AI-human hybrid workflow. The first phase consisted of AOP-helpFinder searching and preliminary analysis. Search terms were developed for each event in the pathway, including KE names, common synonyms, endpoint terms, assay terms, taxonomic descriptors, and relevant species names. These terms were used in AOP-helpFinder to search PubMed for co-occurrence patterns between key events and related mechanistic concepts, following published approaches for literature mining in support of AOP development (Carvaillo et al., 2019; Jornod et al., 2022). The exported results included PMIDs, titles, abstracts, and matched key-event terms. These outputs were subjected to overlap analysis to remove redundant records and to filter the initial literature pool, including elimination of clearly irrelevant records and separation of taxa-related evidence where needed.
The second phase consisted of ChatGPT (OpenAI, San Francisco, CA, USA)-assisted screening. Titles and abstracts from AOP-helpFinder, together with records identified from targeted manual searches, were pre-screened using an large language model (LLM) as an auxiliary prioritization tool. The purpose of this step was not to replace expert judgment, but to increase efficiency and consistency during early evidence triage. The LLM was used to extract study metadata, including stressor, species, biological system, dose or concentration, and exposure duration; identify the evidence type represented in each study, such as biological plausibility, empirical support, or essentiality; and flag weight-of-evidence indicators including dose-response concordance, temporal concordance, incidence concordance, and intervention or rescue evidence. Studies were provisionally classified as high relevance, medium relevance, or low relevance.
High-relevance studies were retrieved for full-text review, whereas medium-relevance studies were retained as potential supporting evidence. Low-relevance studies were documented as low priority and excluded from detailed curation. For studies moved forward to full-text review, a second LLM-assisted pass was used to organize relevant information from the full paper. All LLM-generated outputs were checked directly against the original article text by human reviewers. The LLM was therefore used only as a screening and structuring aid, not as an independent evaluator of the evidence.
Data and methods availability: The literature evidence triage records, including AOP-helpFinder search term libraries, ChatGPT (OpenAI, San Francisco, CA, USA) screening outputs, and human reviewer verification notes, are available from the corresponding author (you.song@niva.no) upon request. All ChatGPT-assisted screening outputs were verified against the original article text by at least one human expert reviewer before any evidence was accepted into KER evidence tables. ChatGPT was used only as an auxiliary prioritization and metadata-extraction tool and not as an independent evaluator of scientific content. The final snapshot PDF of this AOP and the review process PDF will be deposited at the AOP-Wiki forum following completion of the coaching compliance check.
The final phase consisted of manual expert curation and weight-of-evidence evaluation. Domain experts verified the relevance and interpretation of the literature selected in the earlier stages, resolved ambiguous cases, and extracted information to populate KER evidence tables. These tables captured the biological system studied, stressor, method, endpoint, result, concordance pattern, weight-of-evidence category, and bibliographic source. Expert review was then used to evaluate the evidence for each KER in terms of biological plausibility, empirical support, essentiality, quantitative understanding, and evidence gaps. Targeted manual searches were also used to fill specific gaps for ROS biology, oxidative DNA damage, DNA repair, DNA strand breaks, DNA damage response, cell cycle disruption, cell proliferation, and growth inhibition. Studies were prioritized when they measured two or more KEs in the same biological system, reported exposure time and dose or concentration, or provided evidence relevant to dose-response, temporal, or incidence concordance. Mechanistic reviews and OECD reports were used primarily to support biological plausibility, whereas primary experimental studies were prioritized for empirical support wherever possible (Cooke et al., 2003; Cuddihy and O'Connell, 2003; Qian et al., 2009; Hlavová et al., 2011; Quevedo et al., 2021; OECD, 2023).
Summary of the AOP
Events
Molecular Initiating Events (MIE), Key Events (KE), Adverse Outcomes (AO)
| Sequence | Type | Event ID | Title | Short name |
|---|---|---|---|---|
| MIE | 1115 | Increase, Reactive oxygen species | Increase, ROS | |
| KE | 1392 | Increase, Oxidative Stress | Increase, Oxidative Stress | |
| KE | 1634 | Increase, Oxidative DNA damage | Increase, Oxidative DNA damage | |
| KE | 155 | Inadequate DNA repair | Inadequate DNA repair | |
| KE | 1635 | Increase, DNA strand breaks | Increase, DNA strand breaks | |
| KE | 1505 | Increase, Cell cycle disruption | Cell cycle disruption | |
| KE | 1821 | Decrease, Cell proliferation | Decrease, Cell proliferation | |
| AO | 1521 | Decrease, Growth | Decrease, Growth |
Key Event Relationships
| Upstream Event | Relationship Type | Downstream Event | Evidence | Quantitative Understanding |
|---|---|---|---|---|
| Increase, Reactive oxygen species | adjacent | Increase, Oxidative Stress | High | Moderate |
| Increase, Oxidative Stress | adjacent | Increase, Oxidative DNA damage | High | Moderate |
| Increase, Oxidative DNA damage | adjacent | Inadequate DNA repair | High | Low |
| Inadequate DNA repair | adjacent | Increase, DNA strand breaks | High | Low |
| Increase, DNA strand breaks | adjacent | Increase, Cell cycle disruption | High | Moderate |
| Increase, Cell cycle disruption | adjacent | Decrease, Cell proliferation | High | Moderate |
| Decrease, Cell proliferation | adjacent | Decrease, Growth | High | Moderate |
Stressors
| Name | Evidence |
|---|---|
| Hydrogen peroxide | |
| Paraquat | |
| tert-Butyl hydroperoxide | |
| Ionizing Radiation | |
| Ultraviolet B radiation | |
| Silver | |
| Silver nanoparticles | |
| Heavy metals (cadmium, lead, copper, iron, nickel) |
Overall Assessment of the AOP
The overall weight of evidence (WoE) supporting AOP 324 is considered moderate. Biological plausibility is high for all seven KERs in the pathway, reflecting well-established mechanistic connections between ROS accumulation, oxidative stress, oxidative DNA damage, inadequate DNA repair, DNA strand break formation, cell cycle checkpoint activation, reduced cell proliferation, and decreased growth. This biological coherence is reinforced by the reuse of curated KEs and KERs from OECD-endorsed AOPs 263, 296, 478, and 212, which independently support each module in the pathway. Empirical support is high for the upstream (ROSàoxidative stress, oxidative stressàDNA damage relationships), and decreases progressively toward the downstream cell cycle disruption and reduced cell proliferation events, where cross-KE studies in the same biological system are less common. Essentiality of the KEs is rated moderate for most events, reflecting mechanistic support and some intervention evidence but the absence of fully selective, pathway-specific blocking experiments across the whole AOP. Quantitative understanding is low to moderate for most KERs, with the exception of the cell proliferationàgrowth relationship reused from AOP 263, which has moderate quantitative support. The main uncertainties include the relative contribution of the genotoxic route compared with other branches of the broader ROS-growth AOP network, the dual role of ROS in physiological signaling versus toxicological damage, and the multifactorial nature of growth as an apical endpoint. Given the current state of evidence, AOP 324 is most suitable for qualitative and semi-quantitative applications, including mechanistic interpretation of oxidative DNA-damage-driven growth impairment, chemical prioritization, and support for integrated approaches to testing and assessment (IATA). More quantitative regulatory applications will require further empirical studies measuring multiple KEs in the same biological system and under the same stressor exposure (OECD, 2018; Becker et al., 2015).
Domain of Applicability
Life Stage Applicability| Life Stage | Evidence |
|---|---|
| All life stages | Moderate |
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| green algae | Ulva compressa | High | NCBI |
| fish | fish | High | NCBI |
| mammals | mammals | High | NCBI |
| humans | Homo sapiens | High | NCBI |
| crustaceans | Daphnia magna | Moderate | NCBI |
| Sex | Evidence |
|---|---|
| Unspecific | High |
The biological domain of applicability of AOP 324 is defined by the conservation of the KEs and KERs linking oxidative DNA damage to impaired proliferation and growth. The AOP is applicable to aerobic eukaryotic systems in which ROS can oxidize DNA, DNA repair and checkpoint responses regulate genomic integrity, cell cycle progression determines proliferation, and proliferation contributes to growth. The AOP is most relevant to developmental or actively growing systems, including algae, embryos, larvae, juveniles, and proliferative tissues.
The stressor domain is broad but not unlimited. Stressors are relevant when they generate ROS, impair antioxidant defenses, induce oxidative DNA lesions, or produce DNA strand breaks through mechanisms that converge on oxidative damage and checkpoint activation. Examples include redox-cycling organic chemicals, peroxides, metals, nanoparticles, ionizing radiation, ultraviolet radiation, and other stressors capable of producing oxidative DNA damage. The AOP should not be used where decreased growth is driven primarily by mechanisms unrelated to oxidative DNA damage or cell-cycle disruption.
Essentiality of the Key Events
Essentiality was evaluated at the AOP level by considering whether modification of an upstream KE would be expected to prevent, attenuate, or alter downstream KEs and/or the AO. Because many KEs in this AOP represent conserved cellular stress-response processes, direct essentiality evidence is strongest for some biological modules and weaker for others. Evidence is summarized below.
|
Key event |
Essentiality |
Rationale |
Experimental manipulation evidence (KE knock-out / inhibition / rescue) |
Uncertainties |
|
Event 1115: Reactive oxygen species, increased |
Moderate |
ROS are causally linked to oxidative stress because oxidative stress occurs when oxidant formation exceeds antioxidant capacity. Antioxidant and radical-scavenging interventions can reduce oxidative stress and oxidative DNA damage in many systems, supporting the importance of ROS as an upstream driver (Schieber and Chandel, 2014; Sies et al., 2017; OECD, 2023). |
Indirect: antioxidant and ROS-scavenger pre-treatment reduces oxidative stress and downstream damage across oxidative-stress models (Schieber and Chandel, 2014; Sies et al., 2017). No selective single-source ROS knock-out is available. |
ROS can also function in physiological signaling at low levels; oxidative stress can be sustained by altered antioxidant capacity even when a specific ROS source is removed. |
|
Event 1392: Oxidative stress, increased |
High |
Oxidative stress provides the biochemical context for oxidative DNA damage. Excess ROS and redox imbalance can oxidize DNA bases and promote strand breaks; AOP 478 and AOP 296 both use oxidative stress/oxidative DNA damage relationships as central components of endorsed AOP logic (AOP-Wiki, 2026a, 2026b; Cooke et al., 2003; Carrothers et al., 2025; OECD, 2023). |
Indirect: modulation of antioxidant capacity alters progression to oxidative macromolecular damage; oxidative stress is the curated hub KE in endorsed AOP 478 (AOP-Wiki, 2026a; Carrothers et al., 2025). |
Cellular antioxidant and DNA repair capacity can delay or prevent progression to downstream events. |
|
Event 1634: Oxidative DNA damage, increased |
Moderate to high |
AOP 296 identifies oxidative DNA damage as a core initiating event for irreversible genomic damage and reports support from studies using ROS scavengers and DNA repair modulation (AOP-Wiki, 2026b; OECD, 2023). Oxidative lesions such as 8-oxo-dG are mechanistically linked to repair demand and strand break formation (Cooke et al., 2003). |
Indirect: ROS-scavenger and DNA-repair-modulation studies referenced in endorsed AOP 296 alter oxidative DNA lesion burden (AOP-Wiki, 2026b; OECD, 2023; Cooke et al., 2003). |
Direct intervention studies that isolate oxidative DNA damage while keeping other ROS-mediated damage constant are limited. |
|
Event 155: Inadequate DNA repair, increased |
Moderate |
DNA repair capacity determines whether oxidative lesions are correctly resolved or persist as repair intermediates and strand breaks. AOP 296 explicitly includes inadequate DNA repair as a key event connecting oxidative DNA damage to downstream genomic damage (AOP-Wiki, 2026b; OECD, 2023). |
Indirect: repair-capacity modulation changes strand-break persistence; included as a KE in endorsed AOP 296 (AOP-Wiki, 2026b; OECD, 2023). |
Repair systems differ by lesion type, cell-cycle phase, species, and tissue; direct essentiality evidence is context-specific. |
|
Event 1635: DNA strand breaks, increased |
Moderate |
DNA strand breaks are strong activators of DNA damage checkpoint signaling and are central to the transition from molecular damage to cell-cycle disruption. AOP 296 and AOP 478 both include DNA strand breaks in genotoxic pathways downstream of oxidative stress or oxidative DNA damage (AOP-Wiki, 2026a, 2026b; Carrothers et al., 2025; OECD, 2023). |
Indirect: strand-break burden tracks with checkpoint activation; shared with endorsed AOPs 296 and 478 (AOP-Wiki, 2026a, 2026b; OECD, 2023). |
DNA strand breaks may arise from direct DNA attack, repair intermediates, or replication stress; separating these routes can be difficult. |
|
Event 1505: Cell cycle disruption, increased |
Moderate |
Cell-cycle disruption is essential for translating DNA damage into reduced proliferation. AOP 212 reuses cell-cycle disruption as a key event in an endorsed pathway, and DNA damage checkpoint literature supports the causal role of checkpoint activation in delaying or arresting cell-cycle progression (AOP-Wiki, 2026c; Cuddihy and O'Connell, 2003). |
Indirect: checkpoint activation following DNA damage demonstrated in algae and zebrafish cells (Hlavóvá et al., 2011; Quevedo et al., 2021); KE reused from endorsed AOP 212 (AOP-Wiki, 2026c). |
Transient arrest may allow repair and recovery; sustained arrest is more clearly linked to decreased proliferation. |
|
Event 1821: Cell proliferation, decreased |
Moderate |
Reduced proliferation is a direct determinant of tissue and organismal growth. AOP 263 reuses this KE and KER 2205 as the final step from decreased proliferation to decreased growth (AOP-Wiki, 2026d; OECD, 2022; Song and Villeneuve, 2021). |
Indirect: proliferation deficit links bioenergetic/genotoxic upstream to growth; reused from endorsed AOP 263 with KER 2205 (AOP-Wiki, 2026d; Conlon and Raff, 1999; OECD, 2022; Song and Villeneuve, 2021). |
Growth is multifactorial and can also be influenced by energy allocation, nutrition, endocrine signaling, and cell death. |
|
Event 1521: Growth, decreased (AO) |
Not applicable (AO) |
As the adverse outcome, essentiality is assessed for upstream KEs; AOP 263 provides precedent for decreased growth as an AO downstream of these modules (OECD, 2022; Song and Villeneuve, 2021). |
Not applicable (AO). |
|
Weight of Evidence Summary
The evidence assessment is organized by KER. Calls follow the OECD framework for biological plausibility, empirical support, and quantitative understanding (OECD, 2018, 2021).
Biological plausibility of KERs
|
KER |
Evidence |
Rationale |
|
Relationship 2009: ROS increase leads to oxidative stress increase |
High |
The relationship is mechanistic and widely accepted: oxidative stress reflects an imbalance between oxidants and antioxidant defenses, and ROS are the dominant oxidant species represented in this AOP. AOP 478 also describes deposition of energy leading to ROS production and oxidative stress (Schieber and Chandel, 2014; Sies et al., 2017; AOP-Wiki, 2026a; Carrothers et al., 2025). |
|
Relationship 2810: oxidative stress increase leads to oxidative DNA damage increase |
High |
ROS generated under oxidative stress can oxidize DNA bases and damage the sugar-phosphate backbone. This KER is shared with endorsed AOP 478 and provides the upstream connection into the oxidative DNA damage module of AOP 296 (AOP-Wiki, 2026a, 2026b; Cooke et al., 2003; OECD, 2023). |
|
Relationship 1909: oxidative DNA damage increase leads to inadequate DNA repair increase |
High |
Oxidative DNA lesions increase demand on base excision repair and related repair processes. When lesion burden or repair intermediates exceed repair capacity, repair becomes inadequate. This relationship is used in AOP 296 and AOP 478 (AOP-Wiki, 2026a, 2026b; OECD, 2023). |
|
Relationship 1910: inadequate DNA repair increase leads to DNA strand breaks increase |
Moderate to high |
Inadequate or incomplete repair can generate or allow persistence of strand breaks, including repair intermediates and replication-associated breaks. AOP 296 includes this relationship as part of the oxidative DNA damage module (AOP-Wiki, 2026b; OECD, 2023). |
|
Relationship 3480: DNA strand breaks increase leads to cell cycle disruption increase |
High |
DNA strand breaks activate DNA damage response pathways, including ATM/ATR and checkpoint signaling, that delay or arrest cell-cycle progression to allow repair or prevent propagation of damage (Cuddihy and O'Connell, 2003; OECD, 2023). |
|
Relationship 3363: cell cycle disruption increase leads to cell proliferation decrease |
High |
Cell proliferation requires orderly progression through the cell cycle. Sustained checkpoint activation, G1/S arrest, G2/M arrest, or mitotic disruption reduces the rate of cell division. KE 1505 is shared with AOP 212, supporting its reuse as a modular checkpoint-related KE (AOP-Wiki, 2026c; Cuddihy and O'Connell, 2003). |
|
Relationship 2205: cell proliferation decrease leads to growth decrease |
High |
Organismal and tissue growth depend on net accumulation of cell number, cell size, and extracellular components. This relationship is reused from endorsed AOP 263, where decreased cell proliferation is linked to decreased growth (AOP-Wiki, 2026d; Conlon and Raff, 1999; OECD, 2022; Song and Villeneuve, 2021). |
Empirical support for KERs
|
KER |
Evidence |
Rationale |
Inconsistencies or limitations |
|
Relationship 2009: ROS increase leads to oxidative stress increase |
High |
Multiple experimental systems show concordance between ROS generation and oxidative stress biomarkers. For example, paraquat increased ROS and antioxidant enzyme responses in Chlorella vulgaris (Qian et al., 2009), and radiation-induced ROS/oxidative stress relationships are documented in AOP 478 (AOP-Wiki, 2026a; Carrothers et al., 2025). |
Direct ROS measurements are technically challenging and probe-specific; some studies infer ROS from antioxidant responses or downstream damage. |
|
Relationship 2810: oxidative stress increase leads to oxidative DNA damage increase |
Moderate to high |
Oxidative stress is widely associated with oxidative DNA lesions and DNA strand-break endpoints. AOP 478 includes oxidative stress leading to oxidative DNA damage and reports high biological plausibility, while AOP 296 provides a curated oxidative DNA damage module (AOP-Wiki, 2026a, 2026b; Cooke et al., 2003; OECD, 2023). |
Some studies measure DNA strand breaks rather than lesion-specific oxidative DNA damage; stressor-specific pathways may include direct genotoxicity. |
|
Relationship 1909: oxidative DNA damage increase leads to inadequate DNA repair increase |
Moderate |
AOP 296 identifies this relationship and supports it with evidence that excessive oxidative lesions can overwhelm or alter repair processes (AOP-Wiki, 2026b; OECD, 2023). |
Direct co-measurement of oxidative lesions and inadequate repair in the same study is less common than measurement of damage endpoints alone. |
|
Relationship 1910: inadequate DNA repair increase leads to DNA strand breaks increase |
Moderate |
AOP 296 includes inadequate repair leading to DNA strand breaks and broader genomic damage, supported by genotoxicity evidence and repair biology (AOP-Wiki, 2026b; OECD, 2023). |
Directionality can be difficult because strand breaks can both result from repair intermediates and trigger repair responses. |
|
Relationship 3480: DNA strand breaks increase leads to cell cycle disruption increase |
Moderate |
Green algal studies show DNA strand breaks and cell-cycle disruption or division impairment following genotoxic stressors: N-OH-2-AAF-induced DNA strand breaks in Chlamydomonas reinhardtii (David et al., 2009) and zeocin/FdUrd-associated cell-cycle effects in Scenedesmus quadricauda (Hlavová et al., 2011). Silver exposure in embryonic zebrafish cells produced DNA damage and cell-cycle arrest responses (Quevedo et al., 2021). |
Evidence is taxonomically diverse but not always tied specifically to oxidative DNA damage as the upstream cause. |
|
Relationship 3363: cell cycle disruption increase leads to cell proliferation decrease |
Moderate |
Cell-cycle disruption is empirically associated with reduced division and proliferation. AOP 212 provides support for cell-cycle disruption as a reusable KE in an endorsed AOP context, and algal and zebrafish cell studies show reduced division/proliferation following DNA damage-related cell-cycle effects (AOP-Wiki, 2026c; Hlavová et al., 2011; Quevedo et al., 2021). |
Recovery can occur if damage is repaired; transient checkpoint activation does not necessarily lead to long-term proliferation decrease. |
|
Relationship 2205: cell proliferation decrease leads to growth decrease |
Moderate |
AOP 263 reports this relationship as the final growth-relevant KER and assesses it in an endorsed AOP context. Growth effects in algae exposed to paraquat and other stressors provide additional organism-level consistency (AOP-Wiki, 2026d; Qian et al., 2009; Jamers and De Coen, 2010; OECD, 2022; Song and Villeneuve, 2021). |
Growth is an apical endpoint influenced by many processes; direct co-measurement of cell proliferation and organismal growth is less common than measurement of growth alone. |
Inconsistencies and uncertainties
The main uncertainty in AOP 324 is that ROS-mediated growth inhibition can proceed through multiple branches, including genotoxic, energetic, lipid peroxidation, protein oxidation, and cell death pathways. The present AOP captures only the genotoxic branch. Therefore, failure to observe one KE in this linear pathway does not exclude ROS-mediated growth inhibition through another branch of the broader AOP network. Another uncertainty is the dual biological role of ROS. Low or transient ROS levels may support adaptive redox signaling, whereas sustained or excessive ROS promotes oxidative stress and damage. Quantitative thresholds separating adaptive from adverse ROS signaling are not well established across taxa.
A further uncertainty is the directionality and specificity of DNA repair-related events. Oxidative DNA damage can lead to repair activation, repair intermediates, strand breaks, or mutation depending on lesion type and cell-cycle phase. Similarly, DNA strand breaks can be both a consequence of inadequate repair and a stimulus for repair. For AOP-Wiki purposes, the KER sequence used in this AOP follows the linear representation shown in Figure 1 and aligns with the curated oxidative DNA damage module of AOP 296. Finally, the linkage between reduced cell proliferation and decreased growth is biologically strong, but growth is affected by many other physiological factors. This limits quantitative prediction of organismal growth from upstream molecular events.
Quantitative Consideration
Quantitative understanding of AOP 324 is strongest for local relationships that have well-defined biological or assay endpoints, and weaker for full-pathway prediction from ROS increase to decreased growth. Direct quantitative prediction is limited by the short half-life and assay dependence of ROS measurements, variability in oxidative DNA damage endpoints, differences in repair capacity, and the multifactorial nature of growth.
|
KER |
Evidence |
Rationale |
|
Relationship 2009: ROS increase leads to oxidative stress increase |
Low to moderate |
ROS and oxidative stress biomarkers can be measured quantitatively, but direct ROS measurement is probe- and context-dependent. AOP 478 reports high quantitative understanding for deposition of energy leading to oxidative stress, but this does not translate directly into a generalized ROS-to-oxidative-stress response-response model for all stressors (AOP-Wiki, 2026a; Sies et al., 2017). |
|
Relationship 2810: oxidative stress increase leads to oxidative DNA damage increase |
Low to moderate |
Oxidative DNA damage endpoints such as 8-oxo-dG and comet assay endpoints can be quantified, but quantitative prediction from oxidative stress biomarkers to DNA lesion burden remains limited and context-dependent (Cooke et al., 2003; OECD, 2023; AOP-Wiki, 2026a). |
|
Relationship 1909: oxidative DNA damage increase leads to inadequate DNA repair increase |
Low |
Quantitative thresholds at which oxidative DNA damage overwhelms repair capacity are not well generalized across taxa, cell types, or lesion types (OECD, 2023). |
|
Relationship 1910: inadequate DNA repair increase leads to DNA strand breaks increase |
Low |
Repair kinetics can be measured, but quantitative prediction of strand break accumulation from inadequate repair remains context-specific (OECD, 2023). |
|
Relationship 3480: DNA strand breaks increase leads to cell cycle disruption increase |
Moderate |
DNA strand breaks and cell-cycle phase distribution can be quantified, and threshold-like checkpoint responses are biologically expected. However, quantitative relationships vary by cell type, repair capacity, and checkpoint status (Cuddihy and O'Connell, 2003; Hlavová et al., 2011; Quevedo et al., 2021). |
|
Relationship 3363: cell cycle disruption increase leads to cell proliferation decrease |
Moderate |
Cell-cycle arrest and proliferation can both be quantified, but quantitative translation depends on arrest duration, reversibility, and cell population dynamics (Cuddihy and O'Connell, 2003). |
|
Relationship 2205: cell proliferation decrease leads to growth decrease |
Moderate |
AOP 263 reports moderate quantitative understanding for this KER. Growth models and developmental biology support the relationship, but quantitative prediction across taxa and exposure contexts remains limited (AOP-Wiki, 2026d; Conlon and Raff, 1999; OECD, 2022; Song and Villeneuve, 2021). |
BMD/POD-anchored concordance
The following BMD/POD concordance table provides quantitative anchoring for AOP 324 in line with Handbook section 4C. Algal EC50/LOEC values supply POD magnitudes for the downstream energetic and growth events, and the gamma-Daphnia moPOD ordering (Song et al., 2023) is included as cross-network POD-magnitude context. Values are presented as POD magnitudes, not as a causal re-ordering of KEs.
|
Key event (functional category) |
POD metric |
POD value (units as noted) |
POD ordering |
Source |
|
KE 1771: ATP pool, decreased (Chlamydomonas, paraquat) |
EC50 |
0.34 µM |
upstream of death |
Nestler et al., 2012 |
|
KE 55: Cell death (Chlamydomonas, paraquat) |
EC50 |
~1.0 µM |
downstream of ATP |
Nestler et al., 2012 |
|
AO 1521: Growth, decreased (Chlamydomonas, paraquat) |
EC50 / LOEC |
0.26 µM / 0.1 µM |
apical |
Jamers and De Coen, 2010 |
|
KE 1115: ROS, increased (mROS) |
moPOD (multiomics POD) |
0.4 mGy/h |
1 (most sensitive) |
Song et al., 2023 |
Considerations for Potential Applications of the AOP (optional)
AOP 324 can support mechanistic interpretation of toxicity data where ROS generation, oxidative stress, DNA damage, cell cycle disruption, or reduced proliferation are observed together with growth effects. The AOP may be useful for hazard identification, prioritization of stressors that generate oxidative DNA damage, and organization of evidence in IATA or defined approaches. It can also support chemical grouping or read-across where chemicals share evidence of ROS-mediated DNA damage and downstream checkpoint or proliferation effects.
The AOP also highlights assay opportunities and gaps. Several KEs can be measured using established or standardized methods, including DNA strand breaks using OECD TG 489 and growth using OECD growth-related test guidelines such as TG 201 and TG 210 (OECD, 2011, 2013, 2014). Other KEs, such as ROS increase, oxidative DNA damage, inadequate DNA repair, and cell cycle disruption, are supported by mechanistic assays but may require careful interpretation of assay specificity and biological context. High-throughput or other new approach methodology (NAM) assays measuring oxidative stress responses, DNA damage signaling, cell cycle arrest, and proliferation may be mapped to the AOP to support screening and prioritization.
For regulatory application, the AOP is currently most suitable for qualitative or semi-quantitative use, such as supporting biological plausibility, organizing mechanistic evidence, and identifying data gaps. More quantitative applications, such as prediction of decreased growth from upstream ROS or oxidative DNA damage measurements, will require further development of response-response relationships, better characterization of modulating factors, and additional studies measuring multiple KEs in the same biological system over time.
References
Ankley, G. T., Bennett, R. S., Erickson, R. J., Hoff, D. J., Hornung, M. W., Johnson, R. D., Mount, D. R., Nichols, J. W., Russom, C. L., Schmieder, P. K., Serrano, J. A., Tietge, J. E., & Villeneuve, D. L. (2010). Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environmental Toxicology and Chemistry, 29(3), 730-741. https://doi.org/10.1002/etc.34
AOP-Wiki. (2026a). AOP 478: Deposition of energy leading to occurrence of cataracts. Collaborative Adverse Outcome Pathway Wiki. Accessed 14 May 2026.
AOP-Wiki. (2026b). AOP 296: Oxidative DNA damage leading to chromosomal aberrations and mutations. Collaborative Adverse Outcome Pathway Wiki. Accessed 14 May 2026.
AOP-Wiki. (2026c). AOP 212: Histone deacetylase inhibition leading to testicular atrophy. Collaborative Adverse Outcome Pathway Wiki. Accessed 14 May 2026.
AOP-Wiki. (2026d). AOP 263: Uncoupling of oxidative phosphorylation leading to growth inhibition via decreased cell proliferation. Collaborative Adverse Outcome Pathway Wiki. Accessed 14 May 2026.
Becker, R. A., Ankley, G. T., Edwards, S. W., Kennedy, S. W., Linkov, I., Meek, M. E., Sachana, M., Segner, H., Van der Burg, B., Villeneuve, D. L., Watanabe, H., & Barton-Maclaren, T. S. (2015). Increasing scientific confidence in adverse outcome pathways: Application of tailored Bradford-Hill considerations for evaluating weight of evidence. Regulatory Toxicology and Pharmacology, 72(3), 514-537. https://doi.org/10.1016/j.yrtph.2015.04.004
Carrothers, E., et al. (2025). Adverse Outcome Pathway on Deposition of Energy Leading to Cataracts. OECD Series on Adverse Outcome Pathways, No. 40. OECD Publishing, Paris. https://doi.org/10.1787/5ad6b263-en
Carvaillo, J.-C., Barouki, R., Coumoul, X., & Audouze, K. (2019). Linking bisphenol S to adverse outcome pathways using a combined text mining and systems biology approach. Environmental Health Perspectives, 127(4), 047005. https://doi.org/10.1289/EHP4200
Conlon, I., & Raff, M. (1999). Size control in animal development. Cell, 96(2), 235-244. https://doi.org/10.1016/S0092-8674(00)80563-2
Cooke, M. S., Evans, M. D., Dizdaroglu, M., & Lunec, J. (2003). Oxidative DNA damage: Mechanisms, mutation, and disease. The FASEB Journal, 17(10), 1195-1214. https://doi.org/10.1096/fj.02-0752rev
Cuddihy, A. R., & O'Connell, M. J. (2003). Cell-cycle responses to DNA damage in G2. International Review of Cytology, 222, 99-140. https://doi.org/10.1016/S0074-7696(02)22013-6
Fang, P., Li, X., Zhang, Y., & Wang, Z. (2024). Single and joint bioaccumulation and toxicity of isoproturon and cadmium in green algae (Chlamydomonas reinhardtii). Chemical and Biological Technologies in Agriculture, 11, 106. https://doi.org/10.1186/s40538-024-00617-6
Finkel, T., & Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of aging. Nature, 408(6809), 239-247. https://doi.org/10.1038/35041687
Jamers, A., & De Coen, W. (2010). Effect assessment of the herbicide paraquat on a green alga using differential gene expression and biochemical biomarkers. Environmental Toxicology and Chemistry, 29(4), 893-901. https://doi.org/10.1002/etc.102
Jornod, F., Jaylet, T., Blaha, L., Sarigiannis, D., Tamisier, L., & Audouze, K. (2022). AOP-helpFinder webserver: A tool for comprehensive analysis of the literature to support adverse outcome pathways development. Bioinformatics, 38(4), 1173-1175. https://doi.org/10.1093/bioinformatics/btab750
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochemical Journal, 417(1), 1-13. https://doi.org/10.1042/BJ20081386
OECD. (2011). Test No. 201: Freshwater alga and cyanobacteria, growth inhibition test. OECD Guidelines for the Testing of Chemicals, Section 2. OECD Publishing, Paris. https://doi.org/10.1787/9789264069923-en
OECD. (2013). Test No. 210: Fish, early-life stage toxicity test. OECD Guidelines for the Testing of Chemicals, Section 2. OECD Publishing, Paris. https://doi.org/10.1787/9789264203785-en
OECD. (2014). Test No. 489: In vivo mammalian alkaline comet assay. OECD Guidelines for the Testing of Chemicals, Section 4. OECD Publishing, Paris. https://doi.org/10.1787/9789264224179-en
OECD. (2018). Users' Handbook Supplement to the Guidance Document for Developing and Assessing Adverse Outcome Pathways. OECD Series on Adverse Outcome Pathways, No. 1. OECD Publishing, Paris. https://doi.org/10.1787/5jlv1m9d1g32-en
OECD. (2021). Guidance Document for the Scientific Review of Adverse Outcome Pathways. OECD Series on Testing and Assessment, No. 344. OECD Publishing, Paris.
OECD. (2022). Uncoupling of oxidative phosphorylation leading to growth inhibition via decreased cell proliferation. OECD Series on Adverse Outcome Pathways, No. 28. OECD Publishing, Paris. https://doi.org/10.1787/f20867c1-en
OECD. (2023). Oxidative DNA damage leading to chromosomal aberrations and mutations. OECD Series on Adverse Outcome Pathways, No. 29. OECD Publishing, Paris. https://doi.org/10.1787/399d2c34-en
Qian, H., Chen, W., Sun, L., Jin, Y., Liu, W., & Fu, Z. (2009). Inhibitory effects of paraquat on photosynthesis and the response to oxidative stress in Chlorella vulgaris. Ecotoxicology, 18(5), 537-543. https://doi.org/10.1007/s10646-009-0311-8
Quevedo, A. C., Lynch, I., & Valsami-Jones, E. (2021). Cellular repair mechanisms triggered by exposure to silver nanoparticles and ionic silver in embryonic zebrafish cells. Environmental Science: Nano, 8(9), 2507-2522. https://doi.org/10.1039/D1EN00422K
Schieber, M., & Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Current Biology, 24(10), R453-R462. https://doi.org/10.1016/j.cub.2014.03.034
Sies, H., Berndt, C., & Jones, D. P. (2017). Oxidative stress. Annual Review of Biochemistry, 86, 715-748. https://doi.org/10.1146/annurev-biochem-061516-045037
Song, Y., & Villeneuve, D. L. (2021). AOP report: Uncoupling of oxidative phosphorylation leading to growth inhibition via decreased cell proliferation. Environmental Toxicology and Chemistry, 40(11), 2951-2963. https://doi.org/10.1002/etc.5197
Valko, M., Morris, H., & Cronin, M. T. D. (2005). Metals, toxicity and oxidative stress. Current Medicinal Chemistry, 12(10), 1161-1208. https://doi.org/10.2174/0929867053764635
Hlavová, M., Čížková, M., Vítová, M., Bišová, K., & Zachleder, V. (2011). DNA damage during G2 phase does not affect cell cycle progression of the green alga Scenedesmus quadricauda. PLoS ONE, 6(5), e19626. https://doi.org/10.1371/journal.pone.0019626
Appendix 1
List of MIEs in this AOP
Event: 1115: Increase, Reactive oxygen species
Short Name: Increase, ROS
Event Component
| Process | Object | Action |
|---|---|---|
| reactive oxygen species biosynthetic process | reactive oxygen species | increased |
AOPs Including This Key Event
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| cell |
Organ term
| Organ term |
|---|
| organ |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Vertebrates | Vertebrates | High | NCBI |
| human | Homo sapiens | Moderate | NCBI |
| human and other cells in culture | human and other cells in culture | Moderate | NCBI |
| mouse | Mus musculus | Moderate | NCBI |
| crustaceans | Daphnia magna | High | NCBI |
| Lemna minor | Lemna minor | High | NCBI |
| zebrafish | Danio rerio | High | NCBI |
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
| Mixed | High |
ROS is a normal constituent found in all organisms, lifestages, and sexes.
Key Event Description
Biological State: increased reactive oxygen species (ROS)
Biological compartment: an entire cell -- may be cytosolic, may also enter organelles.
Reactive oxygen species (ROS) are O2- derived molecules that can be both free radicals (e.g. superoxide, hydroxyl, peroxyl, alcoxyl) and non-radicals (hypochlorous acid, ozone and singlet oxygen) (Bedard and Krause 2007; Ozcan and Ogun 2015). ROS production occurs naturally in all kinds of tissues inside various cellular compartments, such as mitochondria and peroxisomes (Drew and Leeuwenburgh 2002; Ozcan and Ogun 2015). Furthermore, these molecules have an important function in the regulation of several biological processes – they might act as antimicrobial agents or triggers of animal gamete activation and capacitation (Goud et al. 2008; Parrish 2010; Bisht et al. 2017).
However, in environmental stress situations (exposure to radiation, chemicals, high temperatures) these molecules have its levels drastically increased, and overly interact with macromolecules, namely nucleic acids, proteins, carbohydrates and lipids, causing cell and tissue damage (Brieger et al. 2012; Ozcan and Ogun 2015).
Reactive oxygen species (ROS) refers to the chemical species superoxide, hydrogen peroxide, and their secondary reactive products. In the biological context, ROS are signaling molecules with important roles in cell energy metabolism, cell proliferation, and fate. Therefore, balancing ROS levels at the cellular and tissue level is an important part of many biological processes. Disbalance, mainly an increase in ROS levels, can cause cell dysfunction and irreversible cell damage.
ROS are produced from both exogenous stressors and normal endogenous cellular processes, such as the mitochondrial electron transport chain (ETC). Inhibition of the ETC can result in the accumulation of ROS. Exposure to chemicals, heavy metal ions, or ionizing radiation can also result in increased production of ROS. Chemicals and heavy metal ions can deplete cellular antioxidants reducing the cell’s ability to control cellular ROS and resulting in the accumulation of ROS. Cellular antioxidants include glutathione (GSH), protein sulfhydryl groups, superoxide dismutase (SOD).
ROS are radicals, ions, or molecules that have a single unpaired electron in their outermost shell of electrons, which can be categorized into two groups: free oxygen radicals and non-radical ROS [Liou et al., 2010].
<Free oxygen radicals>
|
superoxide |
O2·- |
|
hydroxyl radical |
·OH |
|
nitric oxide |
NO· |
|
organic radicals |
R· |
|
peroxyl radicals |
ROO· |
|
alkoxyl radicals |
RO· |
|
thiyl radicals |
RS· |
|
sulfonyl radicals |
ROS· |
|
thiyl peroxyl radicals |
RSOO· |
|
disulfides |
RSSR |
<Non-radical ROS>
|
hydrogen peroxide |
H2O2 |
|
singlet oxygen |
1O2 |
|
ozone/trioxygen |
O3 |
|
organic hydroperoxides |
ROOH |
|
hypochlorite |
ClO- |
|
peroxynitrite |
ONOO- |
|
nitrosoperoxycarbonate anion |
O=NOOCO2- |
|
nitrocarbonate anion |
O2NOCO2- |
|
dinitrogen dioxide |
N2O2 |
|
nitronium |
NO2+ |
|
highly reactive lipid- or carbohydrate-derived carbonyl compounds |
|
Potential sources of ROS include NADPH oxidase, xanthine oxidase, mitochondria, nitric oxide synthase, cytochrome P450, lipoxygenase/cyclooxygenase, and monoamine oxidase [Granger et al., 2015]. ROS are generated through NADPH oxidases consisting of p47phox and p67phox. ROS are generated through xanthine oxidase activation in sepsis [Ramos et al., 2018]. Arsenic produces ROS [Zhang et al., 2011]. Mitochondria-targeted paraquat and metformin mediate ROS production [Chowdhury et al., 2020]. ROS are generated by bleomycin [Lu et al., 2010]. Radiation induces dose-dependent ROS production [Ji et al., 2019].
ROS are generated in the course of cellular respiration, metabolism, cell signaling, and inflammation [Dickinson and Chang 2011; Egea et al. 2017]. Hydrogen peroxide is also made by the endoplasmic reticulum in the course of protein folding. Nitric oxide (NO) is produced at the highest levels by nitric oxide synthase in endothelial cells and phagocytes. NO production is one of the main mechanisms by which phagocytes kill bacteria [Wang et al., 2017]. The other species are produced by reactions with superoxide or peroxide, or by other free radicals or enzymes.
ROS activity is principally local. Most ROS have short half-lives, ranging from nano- to milliseconds, so diffusion is limited, while reactive nitrogen species (RNS) nitric oxide or peroxynitrite can survive long enough to diffuse across membranes [Calcerrada et al. 2011]. Consequently, local concentrations of ROS are much higher than average cellular concentrations, and signaling is typically controlled by colocalization with redox buffers [Dickinson and Chang 2011; Egea et al. 2017].
Although their existence is limited temporally and spatially, ROS interact with other ROS or with other nearby molecules to produce more ROS and participate in a feedback loop to amplify the ROS signal, which can increase RNS. Both ROS and RNS also move into neighboring cells, and ROS can increase intracellular ROS signaling in neighboring cells [Egea et al. 2017].
In the primary event, photoreactive chemicals are excited by the absorption of photon energy. The energy of the photoactivated chemicals transfer to oxygen and then generates the reactive oxygen species (ROS), including superoxide (O2−) via type I reaction and singlet oxygen (1O2) via type II reaction, as principal intermediate species in phototoxic reaction (Foote, 1991, Onoue et al. , 2009).
How it is Measured or Detected
Photocolorimetric assays (Sharma et al. 2017; Griendling et al. 2016) or through commercial kits purchased from specialized companies.
Yuan, Yan, et al., (2013) described ROS monitoring by using H2-DCF-DA, a redox-sensitive fluorescent dye. Briefly, the harvested cells were incubated with H2-DCF-DA (50 µmol/L final concentration) for 30 min in the dark at 37°C. After treatment, cells were immediately washed twice, re-suspended in PBS, and analyzed on a BD-FACS Aria flow cytometry. ROS generation was based on fluorescent intensity which was recorded by excitation at 504 nm and emission at 529 nm.
Lipid peroxidation (LPO) can be measured as an indicator of oxidative stress damage Yen, Cheng Chien, et al., (2013).
Chattopadhyay, Sukumar, et al. (2002) assayed the generation of free radicals within the cells and their extracellular release in the medium by addition of yellow NBT salt solution (Park et al., 1968). Extracellular release of ROS converted NBT to a purple colored formazan. The cells were incubated with 100 ml of 1 mg/ml NBT solution for 1 h at 37 °C and the product formed was assayed at 550 nm in an Anthos 2001 plate reader. The observations of the ‘cell-free system’ were confirmed by cytological examination of parallel set of explants stained with chromogenic reactions for NO and ROS.
On the basis of the pathogenesis of drug-induced phototoxicity, a reactive oxygen species (ROS) assay was proposed to evaluate the phototoxic risk of chemicals. The ROS assay can monitor generation of ROS, such as singlet oxygen and superoxide, from photoirradiated chemicals, and the ROS data can be used to evaluate the photoreactivity of chemicals (Onoue et al. , 2014, Onoue et al. , 2013, Onoue and Tsuda, 2006). The ROS assay is a recommended approach by guidelines to evaluate the phototoxic risk of chemicals (ICH, 2014, PCPC, 2014).
<Direct detection>
Many fluorescent compounds can be used to detect ROS, some of which are specific, and others are less specific.
・ROS can be detected by fluorescent probes such as p-methoxy-phenol derivative [Ashoka et al., 2020].
・Chemiluminescence analysis can detect the superoxide, where some probes have a wider range for detecting hydroxyl radical, hydrogen peroxide, and peroxynitrite [Fuloria et al., 2021].
・ROS in the blood can be detected using superparamagnetic iron oxide nanoparticles (SPION)-based biosensor [Lee et al., 2020].
・Hydrogen peroxide (H2O2) can be detected with a colorimetric probe, which reacts with H2O2 in a 1:1 stoichiometry to produce a bright pink colored product, followed by the detection with a standard colorimetric microplate reader with a filter in the 540-570 nm range.
・The levels of ROS can be quantified using multiple-step amperometry using a stainless steel counter electrode and non-leak Ag|AgCl reference node [Flaherty et al., 2017].
・Singlet oxygen can be measured by monitoring the bleaching of p-nitrosodimethylaniline at 440 nm using a spectrophotometer with imidazole as a selective acceptor of singlet oxygen [Onoue et al., 2014].
<Indirect Detection>
Alternative methods involve the detection of redox-dependent changes to cellular constituents such as proteins, DNA, lipids, or glutathione [Dickinson and Chang 2011; Wang et al. 2013; Griendling et al. 2016]. However, these methods cannot generally distinguish between the oxidative species behind the changes and cannot provide good resolution for the kinetics of oxidative activity.
References
Akai, K., et al. (2004). "Ability of ferric nitrilotriacetate complex with three pH-dependent conformations to induce lipid peroxidation." Free Radic Res. Sep;38(9):951-62. doi: 10.1080/1071576042000261945
Ashoka, A. H., et al. (2020). "Recent Advances in Fluorescent Probes for Detection of HOCl and HNO." ACS omega, 5(4), 1730-1742. doi:10.1021/acsomega.9b03420
B.H. Park, S.M. Fikrig, E.M. Smithwick Infection and nitroblue tetrazolium reduction by neutrophils: a diagnostic aid Lancet, 2 (1968), pp. 532-534
Bedard, Karen, and Karl-Heinz Krause. 2007. “The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology.” Physiological Reviews 87 (1): 245–313.
Bisht, Shilpa, Muneeb Faiq, Madhuri Tolahunase, and Rima Dada. 2017. “Oxidative Stress and Male Infertility.” Nature Reviews. Urology 14 (8): 470–85.
Brieger, K., S. Schiavone, F. J. Miller Jr, and K-H Krause. 2012. “Reactive Oxygen Species: From Health to Disease.” Swiss Medical Weekly 142 (August): w13659.
Calcerrada, P., et al. (2011). "Nitric oxide-derived oxidants with a focus on peroxynitrite: molecular targets, cellular responses and therapeutic implications." Curr Pharm Des 17(35): 3905-3932.
Chattopadhyay, Sukumar, et al. "Apoptosis and necrosis in developing brain cells due to arsenic toxicity and protection with antioxidants." Toxicology letters 136.1 (2002): 65-76.
Chowdhury, A. R., et al. (2020). "Mitochondria-targeted paraquat and metformin mediate ROS production to induce multiple pathways of retrograde signaling: A dose-dependent phenomenon." Redox Biol. doi: 10.1016/j.redox.2020.101606. PMID: 32604037; PMCID: PMC7327929.
Dickinson, B. C. and Chang C. J. (2011). "Chemistry and biology of reactive oxygen species in signaling or stress responses." Nature chemical biology 7(8): 504-511.
Drew, Barry, and Christiaan Leeuwenburgh. 2002. “Aging and the Role of Reactive Nitrogen Species.” Annals of the New York Academy of Sciences 959 (April): 66–81.
Egea, J., et al. (2017). "European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS)." Redox biology 13: 94-162.
Flaherty, R. L., et al. (2017). "Glucocorticoids induce production of reactive oxygen species/reactive nitrogen species and DNA damage through an iNOS mediated pathway in breast cancer." Breast Cancer Research, 19(1), 1–13. https://doi.org/10.1186/s13058-017-0823-8
Foote CS. Definition of type I and type II photosensitized oxidation. Photochem Photobiol. 1991;54:659.
Fuloria, S., et al. (2021). "Comprehensive Review of Methodology to Detect Reactive Oxygen Species (ROS) in Mammalian Species and Establish Its Relationship with Antioxidants and Cancer." Antioxidants (Basel, Switzerland) 10(1) 128. doi:10.3390/antiox10010128
Go, Y. M. and Jones, D. P. (2013). "The redox proteome." J Biol Chem 288(37): 26512-26520.
Goud, Anuradha P., Pravin T. Goud, Michael P. Diamond, Bernard Gonik, and Husam M. Abu-Soud. 2008. “Reactive Oxygen Species and Oocyte Aging: Role of Superoxide, Hydrogen Peroxide, and Hypochlorous Acid.” Free Radical Biology & Medicine 44 (7): 1295–1304.
Granger, D. N. and Kvietys, P. R. (2015). "Reperfusion injury and reactive oxygen species: The evolution of a concept" Redox Biol. doi: 10.1016/j.redox.2015.08.020. PMID: 26484802; PMCID: PMC4625011.
Griendling, K. K., et al. (2016). "Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association." Circulation research 119(5): e39-75.
Griendling, Kathy K., Rhian M. Touyz, Jay L. Zweier, Sergey Dikalov, William Chilian, Yeong-Renn Chen, David G. Harrison, Aruni Bhatnagar, and American Heart Association Council on Basic Cardiovascular Sciences. 2016. “Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association.” Circulation Research 119 (5): e39–75.
ICH. ICH Guideline S10 Guidance on Photosafety Evaluation of Pharmaceuticals.: International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2014.
Itziou, A., et al. (2011). "In vivo and in vitro effects of metals in reactive oxygen species production, protein carbonylation, and DNA damage in land snails Eobania vermiculata." Archives of Environmental Contamination and Toxicology, 60(4), 697–707. https://doi.org/10.1007/s00244-010-9583-5
Ji, W. O., et al. "Quantitation of the ROS production in plasma and radiation treatments of biotargets." Sci Rep. 2019 Dec 27;9(1):19837. doi: 10.1038/s41598-019-56160-0. PMID: 31882663; PMCID: PMC6934759.
Kruk, J. and Aboul-Enein, H. Y. (2017). "Reactive Oxygen and Nitrogen Species in Carcinogenesis: Implications of Oxidative Stress on the Progression and Development of Several Cancer Types." Mini-Reviews in Medicinal Chemistry, 17:11. doi:10.2174/1389557517666170228115324
Lee, D. Y., et al. (2020). "PEGylated Bilirubin-coated Iron Oxide Nanoparticles as a Biosensor for Magnetic Relaxation Switching-based ROS Detection in Whole Blood." Theranostics, 10(5), 1997-2007. doi:10.7150/thno.39662
Li, Z., et al. (2020). "Inhibition of MiR-25 attenuates doxorubicin-induced apoptosis, reactive oxygen species production and DNA damage by targeting pten." International Journal of Medical Sciences, 17(10), 1415–1427. https://doi.org/10.7150/ijms.41980
Liou, G. Y. and Storz, P. "Reactive oxygen species in cancer." Free Radic Res. 2010 May;44(5):479-96. doi:10.3109/10715761003667554. PMID: 20370557; PMCID: PMC3880197.
Lu, Y., et al. (2010). "Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production." American journal of respiratory cell and molecular biology, 42(4), 432–441. https://doi.org/10.1165/rcmb.2009-0002OC
Onoue, S., et al. (2013). "Establishment and intra-/inter-laboratory validation of a standard protocol of reactive oxygen species assay for chemical photosafety evaluation." J Appl Toxicol. 33(11):1241-50. doi: 10.1002/jat.2776. Epub 2012 Jun 13. PMID: 22696462.
Onoue S, Hosoi K, Toda T, Takagi H, Osaki N, Matsumoto Y, et al. Intra-/inter-laboratory validation study on reactive oxygen species assay for chemical photosafety evaluation using two different solar simulators. Toxicology in vitro : an international journal published in association with BIBRA. 2014;28:515-23.
Onoue S, Hosoi K, Wakuri S, Iwase Y, Yamamoto T, Matsuoka N, et al. Establishment and intra-/inter-laboratory validation of a standard protocol of reactive oxygen species assay for chemical photosafety evaluation. Journal of applied toxicology : JAT. 2013;33:1241-50.
Onoue S, Kawamura K, Igarashi N, Zhou Y, Fujikawa M, Yamada H, et al. Reactive oxygen species assay-based risk assessment of drug-induced phototoxicity: classification criteria and application to drug candidates. J Pharm Biomed Anal. 2008;47:967-72.
Onoue S, Seto Y, Gandy G, Yamada S. Drug-induced phototoxicity; an early in vitro identification of phototoxic potential of new drug entities in drug discovery and development. Current drug safety. 2009;4:123-36.
Onoue S, Tsuda Y. Analytical studies on the prediction of photosensitive/phototoxic potential of pharmaceutical substances. Pharmaceutical research. 2006;23:156-64.
Ozcan, Ayla, and Metin Ogun. 2015. “Biochemistry of Reactive Oxygen and Nitrogen Species.” In Basic Principles and Clinical Significance of Oxidative Stress, edited by Sivakumar Joghi Thatha Gowder. Rijeka: IntechOpen.
Parrish, A. R. 2010. “2.27 - Hypoxia/Ischemia Signaling.” In Comprehensive Toxicology (Second Edition), edited by Charlene A. McQueen, 529–42. Oxford: Elsevier.
PCPC. PCPC 2014 safety evaluation guidelines; Chapter 7: Evaluation of Photoirritation and Photoallergy potential. Personal Care Products Council; 2014.
Ramos, M. F. P., et al. (2018). "Xanthine oxidase inhibitors and sepsis." Int J Immunopathol Pharmacol. 32:2058738418772210. doi:10.1177/2058738418772210
Ravanat, J. L., et al. (2014). "Radiation-mediated formation of complex damage to DNA: a chemical aspect overview." Br J Radiol 87(1035): 20130715.
Schutzendubel, A. and Polle, A. (2002). "Plant responses to abiotic stresses: heavy metal-induced oxidative stress and protection by mycorrhization." Journal of Experimental Botany, 53(372), 1351–1365. https://doi.org/10.1093/jexbot/53.372.1351
Seto Y, Kato M, Yamada S, Onoue S. Development of micellar reactive oxygen species assay for photosafety evaluation of poorly water-soluble chemicals. Toxicology in vitro : an international journal published in association with BIBRA. 2013;27:1838-46.
Sharma, Gunjan, Nishant Kumar Rana, Priya Singh, Pradeep Dubey, Daya Shankar Pandey, and Biplob Koch. 2017. “p53 Dependent Apoptosis and Cell Cycle Delay Induced by Heteroleptic Complexes in Human Cervical Cancer Cells.” Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie 88 (April): 218–31.
Silva, R., et al. (2019). "Light exposure during growth increases riboflavin production, reactive oxygen species accumulation and DNA damage in Ashbya gossypii riboflavin-overproducing strains." FEMS Yeast Research, 19(1), 1–7. https://doi.org/10.1093/femsyr/foy114
Tsuchiya K, et al. (2005). "Oxygen radicals photo-induced by ferric nitrilotriacetate complex." Biochim Biophys Acta. 1725(1):111-9. doi:10.1016/j.bbagen.2005.05.001
Wang, J., et al. (2017). "Glucocorticoids Suppress Antimicrobial Autophagy and Nitric Oxide Production and Facilitate Mycobacterial Survival in Macrophages." Scientific reports, 7(1), 982. https://doi.org/10.1038/s41598-017-01174-9
Wang, X., et al. (2013). "Imaging ROS signaling in cells and animals." Journal of molecular medicine 91(8): 917-927.
Yen, Cheng Chien, et al. "Inorganic arsenic causes cell apoptosis in mouse cerebrum through an oxidative stress-regulated signaling pathway." Archives of toxicology 85 (2011): 565-575.
Yuan, Yan, et al. "Cadmium-induced apoptosis in primary rat cerebral cortical neurons culture is mediated by a calcium signaling pathway." PloS one 8.5 (2013): e64330.
Zhang, Z., et al. (2011). "Reactive oxygen species mediate arsenic induced cell transformation and tumorigenesis through Wnt/β-catenin pathway in human colorectal adenocarcinoma DLD1 cells. " Toxicology and Applied Pharmacology, 256(2), 114-121. doi:10.1016/j.taap.2011.07.016
List of Key Events in the AOP
Event: 1392: Increase, Oxidative Stress
Short Name: Increase, Oxidative Stress
Event Component
| Process | Object | Action |
|---|---|---|
| oxidative stress | increased |
AOPs Including This Key Event
Stressors
| Name |
|---|
| Acetaminophen |
| Chloroform |
| furan |
| Platinum |
| Aluminum |
| Cadmium |
| Mercury |
| Uranium |
| Arsenic |
| Silver |
| Manganese |
| Nickel |
| Zinc |
| nanoparticles |
Biological Context
| Level of Biological Organization |
|---|
| Molecular |
Domain of Applicability
Taxonomic Applicability Life Stage Applicability| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Taxonomic applicability: Occurrence of oxidative stress is not species specific.
Life stage applicability: Occurrence of oxidative stress is not life stage specific.
Sex applicability: Occurrence of oxidative stress is not sex specific.
Evidence for perturbation by prototypic stressor: There is evidence of the increase of oxidative stress following perturbation from a variety of stressors including exposure to ionizing radiation and altered gravity (Bai et al., 2020; Ungvari et al., 2013; Zhang et al., 2009).
Key Event Description
Oxidative stress is defined as an imbalance in the production of reactive oxygen species (ROS) and antioxidant defenses. High levels of oxidizing free radicals can be very damaging to cells and molecules within the cell. As a result, the cell has important defense mechanisms to protect itself from ROS. For example, Nrf2 is a transcription factor and master regulator of the oxidative stress response. During periods of oxidative stress, Nrf2-dependent changes in gene expression are important in regaining cellular homeostasis (Nguyen, et al., 2009) and can be used as indicators of the presence of oxidative stress in the cell.
In addition to the directly damaging actions of ROS, cellular oxidative stress also changes cellular activities on a molecular level. Redox sensitive proteins have altered physiology in the presence and absence of ROS, which is caused by the oxidation of sulfhydryls to disulfides on neighboring amino acids (Antelmann & Helmann 2011). Importantly Keap1, the negative regulator of Nrf2, is regulated in this manner (Itoh, et al. 2010).
ROS also undermine the mitochondrial defense system from oxidative damage. The antioxidant systems consist of superoxide dismutase, catalase, glutathione peroxidase and glutathione reductase, as well as antioxidants such as α-tocopherol and ubiquinol, or antioxidant vitamins and minerals including vitamin E, C, carotene, lutein, zeaxanthin, selenium, and zinc (Fletcher, 2010). The enzymes, vitamins and minerals catalyze the conversion of ROS to non-toxic molecules such as water and O2. However, these antioxidant systems are not perfect and endogenous metabolic processes and/or exogenous oxidative influences can trigger cumulative oxidative injuries to the mitochondria, causing a decline in their functionality and efficiency, which further promotes cellular oxidative stress (Balasubramanian, 2000; Ganea & Harding, 2006; Guo et al., 2013; Karimi et al., 2017).
However, an emerging viewpoint suggests that ROS-induced modifications may not be as detrimental as previously thought, but rather contribute to signaling processes (Foyer et al., 2017).
Sources of ROS Production
Direct Sources: Direct sources involve the deposition of energy onto water molecules, breaking them into active radical species. When ionizing radiation hits water, it breaks it into hydrogen (H*) and hydroxyl (OH*) radicals by destroying its bonds. The hydrogen will create hydroxyperoxyl free radicals (HO2*) if oxygen is available, which can then react with another of itself to form hydrogen peroxide (H2O2) and more O2 (Elgazzar and Kazem, 2015). Antioxidant mechanisms are also affected by radiation, with catalase (CAT) and peroxidase (POD) levels rising as a result of exposure (Seen et al. 2018; Ahmad et al. 2021).
Indirect Sources: An indirect source of ROS is the mitochondria, which is one of the primary producers in eukaryotic cells (Powers et al., 2008). As much as 2% of the electrons that should be going through the electron transport chain in the mitochondria escape, allowing them an opportunity to interact with surrounding structures. Electron-oxygen reactions result in free radical production, including the formation of hydrogen peroxide (H2O2) (Zhao et al., 2019). The electron transport chain, which also creates ROS, is activated by free adenosine diphosphate (ADP), O2, and inorganic phosphate (Pi) (Hargreaves et al. 2020; Raimondi et al. 2020; Vargas-Mendoza et al. 2021). The first and third complexes of the transport chain are the most relevant to mammalian ROS production (Raimondi et al., 2020). The mitochondria has its own set of DNA and it is a prime target of oxidative damage (Guo et al., 2013). ROS is also produced through nicotinamide adenine dinucleotide phosphate oxidase (Nox) stimulation, an event commenced by angiotensin II, a product/effector of the renin-angiotensin system (Nguyen Dinh Cat et al. 2013; Forrester et al. 2018). Other ROS producers include xanthine oxidase, immune cells (macrophage, neutrophils, monocytes, and eosinophils), phospholipase A2 (PLA2), monoamine oxidase (MAO), and carbon-based nanomaterials (Powers et al. 2008; Jacobsen et al. 2008; Vargas-Mendoza et al. 2021).
How it is Measured or Detected
Oxidative Stress: Direct measurement of ROS is difficult because ROS are unstable. The presence of ROS can be assayed indirectly by measurement of cellular antioxidants, or by ROS-dependent cellular damage. Listed below are common methods for detecting the KE, however there may be other comparable methods that are not listed
- Detection of ROS by chemiluminescence (https://www.sciencedirect.com/science/article/abs/pii/S0165993606001683)
- Detection of ROS by chemiluminescence is also described in OECD TG 495 to assess phototoxic potential.
- Glutathione (GSH) depletion. GSH can be measured by assaying the ratio of reduced to oxidized glutathione (GSH:GSSG) using a commercially available kit (e.g., http://www.abcam.com/gshgssg-ratio-detection-assay-kit-fluorometric-green- ab138881.html).
- TBARS. Oxidative damage to lipids can be measured by assaying for lipid peroxidation using TBARS (thiobarbituric acid reactive substances) using a commercially available kit.
- 8-oxo-dG. Oxidative damage to nucleic acids can be assayed by measuring 8-oxo-dG adducts (for which there are a number of ELISA based commercially available kits),or HPLC, described in Chepelev et al. (Chepelev, et al. 2015).
Molecular Biology: Nrf2. Nrf2’s transcriptional activity is controlled post-translationally by oxidation of Keap1. Assay for Nrf2 activity include:
- Immunohistochemistry for increases in Nrf2 protein levels and translocation into the nucleus Western blot for increased Nrf2 protein levels
- Western blot of cytoplasmic and nuclear fractions to observe translocation of Nrf2 protein from the cytoplasm to the nucleus qPCR of Nrf2 target genes (e.g., Nqo1, Hmox-1, Gcl, Gst, Prx, TrxR, Srxn), or by commercially available pathway-based qPCR array (e.g., oxidative stress array from SABiosciences)
- Whole transcriptome profiling by microarray or RNA-seq followed by pathway analysis (in IPA, DAVID, metacore, etc.) for enrichment of the Nrf2 oxidative stress response pathway (e.g., Jackson et al. 2014)
- OECD TG422D describes an ARE-Nrf2 Luciferase test method
In general, there are a variety of commercially available colorimetric or fluorescent kits for detecting Nrf2 activation.
|
Assay Type & Measured Content |
Description |
Dose Range Studied |
Assay Characteristics (Length/Ease of use/Accuracy) |
|
ROS Formation in the Mitochondria assay (Shaki et al., 2012) |
“The mitochondrial ROS measurement was performed flow cytometry using DCFH-DA. Briefly, isolated kidney mitochondria were incubated with UA (0, 50, 100 and 200 µM) in respiration buffer containing (0.32 mM sucrose, 10mM Tris, 20 mM Mops, 50 µM EGTA, 0.5 mM MgCl2, 0.1 mM KH2PO4 and 5 mM sodium succinate) [32]. In the interval times of 5, 30 and 60 min following the UA addition, a sample was taken and DCFH-DA was added (final concentration, 10 µM) to mitochondria and was then incubated for 10 min.Uranyl acetate-induced ROS generation in isolated kidney mitochondria were determined through the flow cytometry (Partec, Deutschland) equipped with a 488-nm argon ion laser and supplied with the Flomax software and the signals were obtained using a 530-nm bandpass filter (FL-1 channel). Each determination is based on the mean fluorescence intensity of 15,000 counts.”
|
0, 50,100 and 200 µM of Uranyl Acetate
|
Long/ Easy High accuracy
|
|
Mitochondrial Antioxidant Content Assay Measuring GSH content (Shaki et al., 2012)
|
“GSH content was determined using DTNB as the indicator and spectrophotometer method for the isolated mitochondria. The mitochondrial fractions (0.5 mg protein/ml) were incubated with various concentrations of uranyl acetate for 1 h at 30 °C and then 0.1 ml of mitochondrial fractions was added into 0.1 mol/l of phosphate buffers and 0.04% DTNB in a total volume of 3.0 ml (pH 7.4). The developed yellow color was read at 412 nm on a spectrophotometer (UV-1601 PC, Shimadzu, Japan). GSH content was expressed as µg/mg protein.” |
0, 50, 100, or 200 µM Uranyl Acetate |
|
|
H2O2 Production Assay Measuring H2O2 Production in isolated mitochondria (Heyno et al., 2008)
|
“Effect of CdCl2 and antimycin A (AA) on H2O2 production in isolated mitochondria from potato. H2O2 production was measured as scopoletin oxidation. Mitochondria were incubated for 30 min in the measuring buffer (see the Materials and Methods) containing 0.5 mM succinate as an electron donor and 0.2 µM mesoxalonitrile 3‐chlorophenylhydrazone (CCCP) as an uncoupler, 10 U horseradish peroxidase and 5 µM scopoletin.” |
0, 10, 30 µM Cd2+
2 µM antimycin A |
|
|
Flow Cytometry ROS & Cell Viability (Kruiderig et al., 1997)
|
“For determination of ROS, samples taken at the indicated time points were directly transferred to FACScan tubes. Dih123 (10 mM, final concentration) was added and cells were incubated at 37°C in a humidified atmosphere (95% air/5% CO2) for 10 min. At t 5 9, propidium iodide (10 mM, final concentration) was added, and cells were analyzed by flow cytometry at 60 ml/min. Nonfluorescent Dih123 is cleaved by ROS to fluorescent R123 and detected by the FL1 detector as described above for Dc (Van de Water 1995)”“For determination of ROS, samples taken at the indicated time points were directly transferred to FACScan tubes. Dih123 (10 mM, final concentration) was added and cells were incubated at 37°C in a humidified atmosphere (95% air/5% CO2) for 10 min. At t 5 9, propidium iodide (10 mM, final concentration) was added, and cells were analyzed by flow cytometry at 60 ml/min. Nonfluorescent Dih123 is cleaved by ROS to fluorescent R123 and detected by the FL1 detector as described above for Dc (Van de Water 1995)” |
|
Strong/easy medium |
|
DCFH-DA Assay Detection of hydrogen peroxide production (Yuan et al., 2016) |
Intracellular ROS production was measured using DCFH-DA as a probe. Hydrogen peroxide oxidizes DCFH to DCF. The probe is hydrolyzed intracellularly to DCFH carboxylate anion. No direct reaction with H2O2 to form fluorescent production.
|
0-400 µM |
Long/ Easy High accuracy |
|
H2-DCF-DAAssay Detection of superoxide production (Thiebault etal., 2007)
|
This dye is a stable nonpolar compound which diffuses readily into the cells and yields H2-DCF. Intracellular OH or ONOO- react with H2-DCF when cells contain peroxides, to form the highly fluorescent compound DCF, which effluxes the cell. Fluorescence intensity of DCF is measured using a fluorescence spectrophotometer. |
0–600 µM |
Long/ Easy High accuracy |
|
CM-H2DCFDA Assay (Eruslanov & Kusmartsev, 2009) |
The dye (CM-H2DCFDA) diffuses into the cell and is cleaved by esterases, the thiol reactive chlormethyl group reacts with intracellular glutathione which can be detected using flow cytometry. |
|
Long/Easy/ High Accuracy |
|
Method of Measurement |
References |
Description |
OECD-Approved Assay |
|
|
Chemiluminescence |
(Lu, C. et al., 2006; Griendling, K. K., et al., 2016) |
ROS can induce electron transitions in molecules, leading to electronically excited products. When the electrons transition back to ground state, chemiluminescence is emitted and can be measured. Reagents such as luminol and lucigenin are commonly used to amplify the signal. |
No
|
|
|
Spectrophotometry |
(Griendling, K. K., et al., 2016) |
NO has a short half-life. However, if it has been reduced to nitrite (NO2-), stable azocompounds can be formed via the Griess Reaction, and further measured by spectrophotometry. |
No |
|
|
Direct or Spin Trapping-Based electron paramagnetic resonance (EPR) Spectroscopy |
(Griendling, K. K., et al., 2016) |
The unpaired electrons (free radicals) found in ROS can be detected with EPR and is known as electron paramagnetic resonance. A variety of spin traps can be used. |
No |
|
|
Nitroblue Tetrazolium Assay |
(Griendling, K. K., et al., 2016) |
The Nitroblue Tetrazolium assay is used to measure O2.− levels. O2.− reduces nitroblue tetrazolium (a yellow dye) to formazan (a blue dye), and can be measured at 620 nm. |
No |
|
|
Fluorescence analysis of dihydroethidium (DHE) or Hydrocyans |
(Griendling, K. K., et al., 2016) |
Fluorescence analysis of DHE is used to measure O2.− levels. O2.− is reduced to O2 as DHE is oxidized to 2-hydroxyethidium, and this reaction can be measured by fluorescence. Similarly, hydrocyans can be oxidized by any ROS, and measured via fluorescence. |
No |
|
|
Amplex Red Assay |
(Griendling, K. K., et al., 2016) |
Fluorescence analysis to measure extramitochondrial or extracellular H2O2 levels. In the presence of horseradish peroxidase and H2O2, Amplex Red is oxidized to resorufin, a fluorescent molecule measurable by plate reader. |
No |
|
|
Dichlorodihydrofluorescein Diacetate (DCFH-DA) |
(Griendling, K. K., et al., 2016) |
An indirect fluorescence analysis to measure intracellular H2O2 levels. H2O2 interacts with peroxidase or heme proteins, which further react with DCFH, oxidizing it to dichlorofluorescein (DCF), a fluorescent product. |
No |
|
|
HyPer Probe |
(Griendling, K. K., et al., 2016) |
Fluorescent measurement of intracellular H2O2 levels. HyPer is a genetically encoded fluorescent sensor that can be used for in vivo and in situ imaging. |
No |
|
|
Cytochrome c Reduction Assay |
(Griendling, K. K., et al., 2016) |
The cytochrome c reduction assay is used to measure O2.− levels. O O2.− is reduced to O2 as ferricytochrome c is oxidized to ferrocytochrome c, and this reaction can be measured by an absorbance increase at 550 nm. |
No |
|
|
Proton-electron double-resonance imaging (PEDRI) |
(Griendling, K. K., et al., 2016) |
The redox state of tissue is detected through nuclear magnetic resonance/magnetic resonance imaging, with the use of a nitroxide spin probe or biradical molecule. |
No
|
|
|
Glutathione (GSH) depletion |
(Biesemann, N. et al., 2018) |
A downstream target of the Nrf2 pathway is involved in GSH synthesis. As an indication of oxidation status, GSH can be measured by assaying the ratio of reduced to oxidized glutathione (GSH:GSSG) using a commercially available kit (e.g., http://www.abcam.com/gshgssg-ratio-detection-assay-kit-fluorometric-green-ab138881.html). |
No |
|
|
Thiobarbituric acid reactive substances (TBARS) |
(Griendling, K. K., et al., 2016) |
Oxidative damage to lipids can be measured by assaying for lipid peroxidation with TBARS using a commercially available kit. |
No |
|
|
Protein oxidation (carbonylation) |
(Azimzadeh et al., 2017; Azimzadeh et al., 2015; Ping et al., 2020) |
Can be determined with ELISA or a commercial assay kit. Protein oxidation can indicate the level of oxidative stress. |
No |
|
|
Seahorse XFp Analyzer |
Leung et al. 2018 |
The Seahorse XFp Analyzer provides information on mitochondrial function, oxidative stress, and metabolic dysfunction of viable cells by measuring respiration (oxygen consumption rate; OCR) and extracellular pH (extracellular acidification rate; ECAR). |
No |
|
Molecular Biology: Nrf2. Nrf2’s transcriptional activity is controlled post-translationally by oxidation of Keap1. Assays for Nrf2 activity include:
|
Method of Measurement |
References |
Description |
OECD-Approved Assay |
|
Immunohistochemistry |
(Amsen, D., de Visser, K. E., and Town, T., 2009) |
Immunohistochemistry for increases in Nrf2 protein levels and translocation into the nucleus |
No |
|
qPCR |
(Forlenza et al., 2012) |
qPCR of Nrf2 target genes (e.g., Nqo1, Hmox-1, Gcl, Gst, Prx, TrxR, Srxn), or by commercially available pathway-based qPCR array (e.g., oxidative stress array from SABiosciences) |
No |
|
Whole transcriptome profiling via microarray or via RNA-seq followed by a pathway analysis |
(Jackson, A. F. et al., 2014) |
Whole transcriptome profiling by microarray or RNA-seq followed by pathway analysis (in IPA, DAVID, metacore, etc.) for enrichment of the Nrf2 oxidative stress response pathway |
No |
References
Ahmad, S. et al. (2021), “60Co-γ Radiation Alters Developmental Stages of Zeugodacus cucurbitae (Diptera: Tephritidae) Through Apoptosis Pathways Gene Expression”, Journal Insect Science, Vol. 21/5, Oxford University Press, Oxford, https://doi.org/10.1093/jisesa/ieab080
Antelmann, H. and J. D. Helmann (2011), “Thiol-based redox switches and gene regulation.”, Antioxidants & Redox Signaling, Vol. 14/6, Mary Ann Leibert Inc., Larchmont, https://doi.org/10.1089/ars.2010.3400
Amsen, D., de Visser, K. E., and Town, T. (2009), “Approaches to determine expression of inflammatory cytokines”, in Inflammation and Cancer, Humana Press, Totowa, https://doi.org/10.1007/978-1-59745-447-6_5
Azimzadeh, O. et al. (2015), “Integrative Proteomics and Targeted Transcriptomics Analyses in Cardiac Endothelial Cells Unravel Mechanisms of Long-Term Radiation-Induced Vascular Dysfunction”, Journal of Proteome Research, Vol. 14/2, American Chemical Society, Washington, https://doi.org/10.1021/pr501141b
Azimzadeh, O. et al. (2017), “Proteome analysis of irradiated endothelial cells reveals persistent alteration in protein degradation and the RhoGDI and NO signalling pathways”, International Journal of Radiation Biology, Vol. 93/9, Informa, London, https://doi.org/10.1080/09553002.2017.1339332
Azzam, E. I. et al. (2012), “Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury”, Cancer Letters, Vol. 327/1-2, Elsevier, Ireland, https://doi.org/10.1016/j.canlet.2011.12.012
Bai, J. et al. (2020), “Irradiation-induced senescence of bone marrow mesenchymal stem cells aggravates osteogenic differentiation dysfunction via paracrine signaling”, American Journal of Physiology - Cell Physiology, Vol. 318/5, American Physiological Society, Rockville, https://doi.org/10.1152/ajpcell.00520.2019.
Balasubramanian, D (2000), “Ultraviolet radiation and cataract”, Journal of ocular pharmacology and therapeutics, Vol. 16/3, Mary Ann Liebert Inc., Larchmont, https://doi.org/10.1089/jop.2000.16.285.
Biesemann, N. et al., (2018), “High Throughput Screening of Mitochondrial Bioenergetics in Human Differentiated Myotubes Identifies Novel Enhancers of Muscle Performance in Aged Mice”, Scientific Reports, Vol. 8/1, Nature Portfolio, London, https://doi.org/10.1038/s41598-018-27614-8.
Elgazzar, A. and N. Kazem. (2015), “Chapter 23: Biological effects of ionizing radiation” in The Pathophysiologic Basis of Nuclear Medicine, Springer, New York, pp. 540-548
Eruslanov, E., & Kusmartsev, S. (2010). Identification of ROS using oxidized DCFDA and flow-cytometry. Methods in molecular biology ,N.J., Vol. 594, https://doi.org/10.1007/978-1-60761-411-1_4
Fletcher, A. E (2010), “Free radicals, antioxidants and eye diseases: evidence from epidemiological studies on cataract and age-related macular degeneration”, Ophthalmic Research, Vol. 44, Karger International, Basel, https://doi.org/10.1159/000316476.
Forlenza, M. et al. (2012), “The use of real-time quantitative PCR for the analysis of cytokine mRNA levels” in Cytokine Protocols, Springer, New York, https://doi.org/10.1007/978-1-61779-439-1_2
Forrester, S.J. et al. (2018), “Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology”, Physiological Reviews, Vol. 98/3, American Physiological Society, Rockville, https://doi.org/10.1152/physrev.00038.201
Foyer, C. H., A. V. Ruban, and G. Noctor (2017), “Viewing oxidative stress through the lens of oxidative signalling rather than damage”, Biochemical Journal, Vol. 474/6, Portland Press, England, https://doi.org/10.1042/BCJ20160814
Ganea, E. and J. J. Harding (2006), “Glutathione-related enzymes and the eye”, Current eye research, Vol. 31/1, Informa, London, https://doi.org/10.1080/02713680500477347.
Griendling, K. K. et al. (2016), “Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association”, Circulation research, Vol. 119/5, Lippincott Williams & Wilkins, Philadelphia, https://doi.org/10.1161/RES.0000000000000110
Guo, C. et al. (2013), “Oxidative stress, mitochondrial damage and neurodegenerative diseases”, Neural regeneration research, Vol. 8/21, Publishing House of Neural Regeneration Research, China, https://doi.org/10.3969/j.issn.1673-5374.2013.21.009
Hargreaves, M., and L. L. Spriet (2020), “Skeletal muscle energy metabolism during exercise.”, Nature Metabolism, Vol. 2, Nature Portfolio, London, https://doi.org/10.1038/s42255-020-0251-4
Hladik, D. and S. Tapio (2016), “Effects of ionizing radiation on the mammalian brain”, Mutation Research/Reviews in Mutation Research, Vol. 770, Elsevier, Amsterdam, https://doi.org/10.1016/j.mrrev.2016.08.003
Itoh, K., J. Mimura and M. Yamamoto (2010), “Discovery of the negative regulator of Nrf2, Keap1: a historical overview”, Antioxidants & Redox Signaling, Vol. 13/11, Mary Ann Leibert Inc., Larchmont, https://doi.org/10.1089/ars.2010.3222
Jackson, A.F. et al. (2014), “Case study on the utility of hepatic global gene expression profiling in the risk assessment of the carcinogen furan.”, Toxicology and Applied Pharmacology, Vol. 274/11, Elsevier, Amsterdam, https://doi.org/10.1016/j.taap.2013.10.019
Jacobsen, N.R. et al. (2008), “Genotoxicity, cytotoxicity, and reactive oxygen species induced by single-walled carbon nanotubes and C60 fullerenes in the FE1-MutaTM Mouse lung epithelial cells”, Environmental and Molecular Mutagenesis, Vol. 49/6, John Wiley & Sons, Inc., Hoboken, https://doi.org/10.1002/em.20406
Karimi, N. et al. (2017), “Radioprotective effect of hesperidin on reducing oxidative stress in the lens tissue of rats”, International Journal of Pharmaceutical Investigation, Vol. 7/3, Phcog Net, Bengaluru, https://doi.org/10.4103/jphi.JPHI_60_17.
Leung, D.T.H., and Chu, S. (2018), “Measurement of Oxidative Stress: Mitochondrial Function Using the Seahorse System” In: Murthi, P., Vaillancourt, C. (eds) Preeclampsia. Methods in Molecular Biology, vol 1710. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7498-6_22
Lu, C., G. Song, and J. Lin (2006), “Reactive oxygen species and their chemiluminescence-detection methods”, TrAC Trends in Analytical Chemistry, Vol. 25/10, Elsevier, Amsterdam, https://doi.org/10.1016/j.trac.2006.07.007
Nguyen Dinh Cat, A. et al. (2013), “Angiotensin II, NADPH oxidase, and redox signaling in the vasculature”, Antioxidants & redox signaling, Vol. 19/10, Mary Ann Liebert, Larchmont, https://doi.org/10.1089/ars.2012.4641
Ping, Z. et al. (2020), “Oxidative Stress in Radiation-Induced Cardiotoxicity”, Oxidative Medicine and Cellular Longevity, Vol. 2020, Hindawi, https://doi.org/10.1155/2020/3579143
Powers, S.K. and M.J. Jackson. (2008), “Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production”, Physiological Reviews, Vol. 88/4, American Physiological Society, Rockville, https://doi.org/10.1152/physrev.00031.2007
Raimondi, V., F. Ciccarese and V. Ciminale. (2020), “Oncogenic pathways and the electron transport chain: a dangeROS liason”, British Journal of Cancer, Vol. 122/2, Nature Portfolio, London, https://doi.org/10.1038/s41416-019-0651-y
Seen, S. and L. Tong. (2018), “Dry eye disease and oxidative stress”, Acta Ophthalmologica, Vol. 96/4, John Wiley & Sons, Inc., Hoboken, https://doi.org/10.1111/aos.13526
Ungvari, Z. et al. (2013), “Ionizing Radiation Promotes the Acquisition of a Senescence-Associated Secretory Phenotype and Impairs Angiogenic Capacity in Cerebromicrovascular Endothelial Cells: Role of Increased DNA Damage and Decreased DNA Repair Capacity in Microvascular Radiosensitivity”, The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, Vol. 68/12, Oxford University Press, Oxford, https://doi.org/10.1093/gerona/glt057.
Vargas-Mendoza, N. et al. (2021), “Oxidative Stress, Mitochondrial Function and Adaptation to Exercise: New Perspectives in Nutrition”, Life, Vol. 11/11, Multidisciplinary Digital Publishing Institute, Basel, https://doi.org/10.3390/life11111269
Wang, H. et al. (2019), “Radiation-induced heart disease: a review of classification, mechanism and prevention”, International Journal of Biological Sciences, Vol. 15/10, Ivyspring International Publisher, Sydney, https://doi.org/10.7150/ijbs.35460
Zhang, R. et al. (2009), “Blockade of AT1 receptor partially restores vasoreactivity, NOS expression, and superoxide levels in cerebral and carotid arteries of hindlimb unweighting rats”, Journal of applied physiology, Vol. 106/1, American Physiological Society, Rockville, https://doi.org/10.1152/japplphysiol.01278.2007.
Zhao, R. Z. et al. (2019), “Mitochondrial electron transport chain, ROS generation and uncoupling”, International journal of molecular medicine, Vol. 44/1, Spandidos Publishing Ltd., Athens, https://doi.org/10.3892/ijmm.2019.4188
Event: 1634: Increase, Oxidative DNA damage
Short Name: Increase, Oxidative DNA damage
Event Component
| Process | Object | Action |
|---|---|---|
| regulation of response to reactive oxygen species | reactive oxygen species | occurrence |
AOPs Including This Key Event
Stressors
| Name |
|---|
| Hydrogen peroxide |
| Potassium bromate |
| Ionizing Radiation |
| Sodium arsenite |
| Reactive oxygen species |
Biological Context
| Level of Biological Organization |
|---|
| Molecular |
Cell term
| Cell term |
|---|
| eukaryotic cell |
Organ term
| Organ term |
|---|
| organ |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human and other cells in culture | human and other cells in culture | Moderate | NCBI |
| yeast | Saccharomyces cerevisiae | Low | NCBI |
| mouse | Mus musculus | High | NCBI |
| rat | Rattus norvegicus | Low | NCBI |
| bovine | Bos taurus | Low | NCBI |
| human | Homo sapiens | High | NCBI |
| rabbit | Oryctolagus cuniculus | Low | NCBI |
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | Moderate |
Taxonomic applicability: Theoretically, DNA oxidation can occur in any cell type, in any organism. Oxidative DNA lesions have been measured in mammalian cells (human, mouse, calf, rat) in vitro and in vivo, and in prokaryotes.
Life stage applicability: This key event is not life stage specific (Mesa & Bassnett, 2013; Suman et al., 2019).
Sex applicability: This key event is not sex specific (Mesa & Bassnett, 2013).
Evidence for Perturbation by Prototypic Stressor: H2O2 and KBrO3 – A concentration-dependent increase in oxidative lesions was observed in both Fpg- and hOGG1-modified comet assays of TK6 cells treated with increasing concentrations of glucose oxidase (an enzyme that generates H2O2) and potassium bromate for 4 h (Platel et al., 2011).
Evidence indicates that oxidative DNA damage is also induced by X-rays (Bahia et al., 2018), 60Co γ-rays, 12C ions, α particles, electrons (Georgakilas, 2013), UVB (Mesa and Bassnett, 2013), γ-rays, 56Fe ions (Datta et al., 2012), and protons (Suman et al., 2019).
Key Event Description
The nitrogenous bases of DNA are susceptible to oxidation in the presence of oxidizing agents. Oxidative adducts form mainly on C5 and to a lesser degree on C6 of thymine and cytosine, and on C8 of guanine and adenine. Guanine is most prone to oxidation due to its low oxidation potential (Jovanovic and Simic, 1986). Indeed, 8-oxo-2’-deoxyguanosine (8-oxodG)/8-hydroxy-2’-deoxyguanosine (8-OHdG) is the most abundant and well-studied oxidative DNA lesion in the cell (Swenberg et al., 2011). It causes an A(anti):8-oxo-G(syn) mispair instead of the normal C(anti):8-oxo-G(syn) pair. This pairing does not cause large structural changes to the DNA backbone, and therefore remains undetected by the polymerase’s proofreading mechanism. Consequently, one of the daughter strands will have an AT pair instead of the correct GC pair after replication (Markkanen, 2017).
Formamidopyrimidine lesions on guanine and adenine (FaPyG and FaPyA), 8-hydroxy-2'-deoxyadenine (8-oxodA), and thymidine glycol (Tg) are other common oxidative lesions. We refer the reader to reviews on this topic to see the full set of potential oxidative DNA lesions (Whitaker et al., 2017). Oxidative DNA lesions are present in the cell at a steady state due to endogenous redox processes (Swenberg et al., 2010). Under normal conditions, cells are able to withstand the baseline level of oxidized bases through efficient repair and regulation of free radicals in the cell. However, direct chemical insult from specific compounds, exposure to various forms of radiation, or induction of reactive oxygen species (ROS) from the reduction of endogenous molecules, as well as through the release of inflammatory cell-derived oxidants, can lead to increased DNA oxidation, a state known as oxidative stress (Turner et al., 2002; Schoenfeld et al., 2012; Tangvarasittichai and Tangvarasittichai, 2019). It is worth noting that ROS must be generated near the DNA to cause damage, otherwise, if ROS was produced more distantly, then it can be removed by the cell (Nilsson & Liu, 2020). Furthermore, although cells do possess repair mechanisms to deal with oxidative DNA damage, sometimes the repair intermediates can interfere with genome function or decrease stability of the genome. This creates a balancing act between when it is best to repair damage and when it is best to leave it (Poetsch, 2020a).
This KE describes an increase in oxidative lesions of a broad spectrum (ie. superoxide radical (O2•−), hydroxyl radical (OH), peroxyl radical (RO22), single oxygen (1O2 ) in the nuclear DNA above the steady-state level. Oxidative DNA damage can occur in any cell type with nuclear DNA under oxidative stress.
How it is Measured or Detected
Relative Quantification of Oxidative DNA Lesions
- Comet assay (single cell gel electrophoresis) with Fpg and hOGG1 modifications (Smith et al., 2006; Platel et al., 2011)
- Oxoguanine glycosylase (hOGG1) and formamidopyrimidine-DNA glycosylase (Fpg) are base excision repair (BER) enzymes in eukaryotic and prokaryotic cells, respectively
- Both enzymes are bi-functional; the glycosylase function cleaves the glycosidic bond between the ribose and the oxidized base, giving rise to an abasic site, and the apurinic/apymidinic (AP) site lyase function cleaves the phosphodiester bond via β-elimination reaction and creates a single strand break
- Treatment of DNA with either enzyme prior to performing the electrophoresis step of the comet assay allows detection of oxidative lesions by measuring the increase in comet tail length when compared against untreated samples.
- Enzyme-linked immunosorbant assay (ELISA) (Dizdaroglu et al., 2002; Breton et al., 2003; Xu et al., 2008; Zhao et al. 2017)
- 8-oxodG can be detected using immunoassays, such as ELISA, that use antibodies against 8-oxodG lesions. It has been noted that immunodetection of 8-oxodG can be interfered by certain compounds in biological samples.
Absolute Quantification of Oxidative DNA Lesions
- Quantification of 8-oxodG using HPLC-EC (Breton et al., 2003; Chepelev et al., 2015)
- 8-oxodG can be separated from digested DNA and precisely quantified using high performance liquid chromatography (HPLC) with electrochemical detection
- Mass spectrometry LC-MRM/MS (Mangal et al., 2009)
- Liquid chromatography can also be coupled with multiple reaction monitoring/ mass spectrometry to detect and quantify oxidative lesions. Correlation between lesions measured by hOGG1-modified comet assay and LC-MS has been reported
Gas chromatography-mass spectrometry (GC-MS)
- DNA is hydrolyzed to release either free bases or nucleosides and then undergoes derivatization in order to increase their volatility. Finally, samples run through a gas chromatograph and then a mass spectrometer. The mass spectrometer results are used to determine oxidative DNA damage by identifying modified bases or nucleosides (Dizdaroglu, 1994).
Sequencing assays
- Various markers are used to detect and highlight sites of DNA damage; the result is then processed and sequenced. This category encompasses a wide range of assays such as snAP-seq, OGG1-AP-seq, oxiDIP-seq, OG-seq, and click-code-seq (Yun et al., 2017; Wu et al., 2018; Amente et al., 2019; Poetsch, 2020b).
- We note that other types of oxidative lesions can be quantified using the methods described above.
References
Amente, S. et al. (2019), “Genome-wide mapping of 8-oxo-7,8-dihydro-2’-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells”, Nucleic Acids Research 2019, Vol. 47/1, Oxford University Press, England, https://doi.org/10.1093/nar/gky1152
Bahia, S. et al. (2018), “Oxidative and nitrative stress-related changes in human lens epithelial cells following exposure to X-rays”, International journal of radiation biology, Vol. 94/4, England, https://doi.org/10.1080/09553002.2018.1439194
Breton J, Sichel F, Bainchini F, Prevost V. (2003). Measurement of 8-Hydroxy-2′-Deoxyguanosine by a Commercially Available ELISA Test: Comparison with HPLC/Electrochemical Detection in Calf Thymus DNA and Determination in Human Serum. Anal Lett 36:123-134.
Cabrera, M. P., R. Chihuailaf and F. Wittwer Menge (2011), “Antioxidants and the integrity of ocular tissues”, Veterinary medicine international, Vol. 2011, SAGE-Hindawi Access to Research, United States, https://doi.org/10.4061/2011/905153
Cadet, J. et al. (2012), “Oxidatively generated complex DNA damage: tandem and clustered lesions”, Cancer letters, Vol. 327/1, Elsevier Ireland Ltd, Ireland. https://doi.org/10.1016/j.canlet.2012.04.005
Chepelev N, Kennedy D, Gagne R, White T, Long A, Yauk C, White P. (2015). HPLC Measurement of the DNA Oxidation Biomarker, 8-oxo-7,8-dihydro-2'-deoxyguanosine, in Cultured Cells and Animal Tissues. Journal of Visualized Experiments 102:e52697.
Collins, A. R. (2014), “Measuring oxidative damage to DNA and its repair with the comet assay”, Biochimica et biophysica acta. General subjects, Vol. 1840/2, Elsevier B.V., https://doi.org/10.1016/j.bbagen.2013.04.022
Datta, K. et al. (2012), “Exposure to heavy ion radiation induces persistent oxidative stress in mouse intestive”, PloS One, Vol. 7/8, Public Library of Science, United States, https://doi.org/10.1371/journal.pone.0042224
Dizdaroglu, M. (1994), “Chemical determination of oxidative DNA damage by gas chromatography-mass spectrometry”, Methods in Enzymology, Vol. 234, Elsevier Science & Technology, United States, https://doi.org/ 10.1016/0076-6879(94)34072-2
Dizdaroglu, M. et al. (2002), “Free radical-induced damage to DNA : mechanisms and measurement”, Free radical biology & medicine, Vol. 32/11, United States, pp. 1102-1115
Eaton, J. W. (1995), “UV-mediated cataractogenesis: a radical perspective”, Documenta ophthalmologica, Vol. 88/3-4, Springer, Dordrecht, https://doi.org/10.1007/BF01203677
Fletcher, A. E. (2010), “Free radicals, antioxidants and eye diseases: evidence from epidemiological studies on cataract and age-related macular degeneration”, Ophthalmic Research, Vol. 44/3, Karger international, Basel, https://doi.org/10.1159/000316476
Georgakilas, A. G et al. (2013), “Induction and repair of clustered DNA lesions: what do we know so far?”, Radiation Research, Vol. 180/1, The Radiation Research Society, United States, https://doi.org/10.1667/RR3041.1
Jose, D. et al. (2009). “Spectroscopic studies of position-specific DNA “breathing” fluctuations at replication forks and primer-template junctions”, Proceedings of the National Academy of Sciences of the United States of America, Vol. 106/11, https://doi.org/10.1073/pnas.0900803106
Jovanovic S, Simic M. (1986). One-electron redox potential of purines and pyrimidines. J Phys Chem 90:974-978.
Kruk, J., K. Kubasik-Kladna and H. Y. Aboul-Enein (2015), “The role oxidative stress in the pathogenesis of eye diseases: current status and a dual role of physical activity”, Mini-reviews in medicinal chemistry, Vol. 16/3, Bentham Science Publishers Ltd, Netherlands, https://doi.org/10.2174/1389557516666151120114605
Lee, J. et al. (2004), “Reactive oxygen species, aging, and antioxidative nutraceuticals”, Comprehensive reviews in food science and food safety, Vol. 3/1, Blackwell Publishing Ltd, Oxford, https://doi.org/10.1111/j.1541-4337.2004.tb00058.x
Mangal D, Vudathala D, Park J, Lee S, Penning T, Blair I. (2009). Analysis of 7,8-Dihydro-8-oxo-2′-deoxyguanosine in Cellular DNA during Oxidative Stress. Chem Res Toxicol 22:788-797.
Markkanen, E. (2017), “Not breathing is not an option: How to deal with oxidative DNA damage”, DNA repair, Vol. 59, Elsevier B.V., Netherlands, https://doi.org/10.1016/j.dnarep.2017.09.007
Mesa, R. and S. Bassnett (2013), “UV-B induced DNA damage and repair in the mouse lens”, Investigative ophthalmology & visual science, Vol. 54/10, the Association for Research in Vision and Ophthalmology, United States, https://doi.org/10.1167/iovs.13-12644
Nilsson R. and Liu N. (2020), “Nuclear DNA damages generated by reactive oxygen molecules (ROS) under oxidative stress and their relevance to human cancers, including ionizing radiation-induced neoplasia part I: Physical, chemical and molecular biology aspects”, Radiation Medicine and Protection, Vol. 1/3(3), https://doi.org/10.1016/j.radmp.2020.09.002
Pendergrass, W. et al. (2010), “X-ray induced cataract is preceded by LEC loss, and coincident with accumulation of cortical DNA, and ROS; similarities with age-related cataracts”, Molecular vision, Vol. 16, Molecular Vision, United States, pp. 1496-1513
Platel A, Nesslany F, Gervais V, Claude N, Marzin D. (2011). Study of oxidative DNA damage in TK6 human lymphoblastoid cells by use of the thymidine kinase gene-mutation assay and the in vitro modified comet assay: Determination of No-Observed-Genotoxic-Effect-Levels. Mutat Res 726:151-159.
Poetsch, Anna R. (2020a), “The genomics of oxidative DNA damage, repair, and resulting mutagenesis”, Computational and structural biotechnology journal 2020, Vol. 18, Elsevier B.V., Netherlands https://doi.org/10.1016/j.csbj.2019.12.013
Poetsch, A. R. (2020b), “AP-Seq: A method to measure apurinic sites and small base adducts genome-wide”, The Nucleus, Springer US, New York, Sacca, S. C. et al. (2009), “Gene-environment interactions in ocular diseases”, Mutation research – fundamental and molecular mechanisms of mutagenesis, Vol. 667/1-2, Elsevier, Amsterdam, https://doi.org/10.1016/j.mrfmmm.2008.11.002
Schoenfeld, M. P. et al. (2012), “A hypothesis on biological protection from space radiation through the use of new therapeutic gases as medical counter measures”, Medical gas research, Vol. 2/1, BioMed Central Ltd, India, https://doi.org/10.1186/2045-9912-2-8
Smith C, O'Donovan M, Martin E. (2006). hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis 21:185-190.
Stohs, S. J. (1995), “The role of free radicals in toxicity and disease”, Journal of Basic and Clinical Physiology and Pharmacology, Vol. 6/3-4, Freund Publishing House Ltd, https://doi.org/10.1515/JBCPP.1995.6.3-4.205
Suman, S. et al. (2019), “Fractionated and acute proton radiation show differential intestinal tumorigenesis and DNA damage and repair pathway response in ApcMin/+ mice”, International Journal of Radiation Oncology, Biology, Physics, Vol. 105/3, Elsevier Inc, https://doi.org/10.1016/j.ijrobp.2019.06.2532
Swenberg J. et al. (2011). "Endogenous versus Exogenous DNA Adducts: Their Role in Carcinogenesis, Epidemiology, and Risk Assessment." Toxicol Sci 120:S130-S145.
Tangvarasittichai, O and S. Tangvarasittichai (2018), “Oxidative stress, ocular disease, and diabetes retinopathy”, Current Pharmaceutical Design, Vol. 24/40, Bentham Science Publishers, https://doi.org/10.2174/1381612825666190115121531
Turner, N. D. et al. (2002), “Opportunities for nutritional amelioration of radiation-induced cellular damage”, Nutrition, Vol. 18/10, Elsevier Inc, New York, https://doi.org/10.1016/S0899-9007(02)00945-0
Whitaker A, Schaich M, Smith MS, Flynn T, Freudenthal B. (2017). Base excision repair of oxidative DNA damage: from mechanism to disease. Front Biosci 22:1493-1522.
Wu, J. (2018), “Nucleotide-resolution genome-wide mapping of oxidative DNA damage by click-code-seq”, Journal of the American Chemical Society 2018, American Chemical Society, United States https://doi-org.proxy.bib.uottawa.ca/10.1021/jacs.8b03715
Xu, X. et al. (2008). “Fluorescence recovery assay for the detection of protein-DNA binding”, Analytical Chemistry, Vol. 80/14, https://doi.org/10.1021/ac8007016
Zhao M, Howard E, Guo Z, Parris A, Yang X. (2017). p53 pathway determines the cellular response to alcohol-induced DNA damage in MCF-7 breast cancer cells. PLoS One 12:e0175121.
Event: 155: Inadequate DNA repair
Short Name: Inadequate DNA repair
Event Component
| Process | Object | Action |
|---|---|---|
| DNA repair | deoxyribonucleic acid | abnormal |
AOPs Including This Key Event
Stressors
| Name |
|---|
| Ionizing Radiation |
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| mouse | Mus musculus | High | NCBI |
| rat | Rattus norvegicus | Moderate | NCBI |
| Syrian golden hamster | Mesocricetus auratus | Moderate | NCBI |
| Homo sapiens | Homo sapiens | High | NCBI |
| cow | Bos taurus | Low | NCBI |
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
The retention of adducts has been directly measured in many different types of eukaryotic somatic cells (in vitro and in vivo). In male germ cells, work has been done on hamsters, rats and mice. The accumulation of mutation and changes in mutation spectrum has been measured in mice and human cells in culture. Theoretically, saturation of DNA repair occurs in every species (prokaryotic and eukaryotic). The principles of this work were established in prokaryotic models. Nagel et al. (2014) have produced an assay that directly measures DNA repair in human cells in culture.
NHEJ is primarily used by vertebrate multicellular eukaryotes, but it also been observed in plants. Furthermore, it has recently been discovered that some bacteria (Matthews et al., 2014) and yeast (Emerson et al., 2016) also use NHEJ. In terms of invertebrates, most lack the core DNA-PKcs and Artemis proteins; they accomplish end joining by using the RA50:MRE11:NBS1 complex (Chen et al., 2001). HR occurs naturally in eukaryotes, bacteria, and some viruses (Bhatti et al., 2016).
Taxonomic applicability: Inadequate DNA repair is applicable to all species, as they all contain DNA (White & Vijg, 2016).
Life stage applicability: This key event is not life stage specific as any life stage can have poor repair, though as individuals age their repair process become less effective (Gorbunova & Seluanov, 2016).
Sex applicability: There is no evidence of sex-specificity for this key event, with initial rate of DNA repair not significantly different between sexes (Trzeciak et al., 2008).
Evidence for perturbation by a stressor: Multiple studies demonstrate that inadequate DNA repair can occur as a result of stressors such as ionizing and non-ionizing radiation, as well as chemical agents (Kuhne et al., 2005; Rydberg et al., 2005; Dahle et al., 2008; Seager et al., 2012; Wilhelm, 2014; O’Brien et al., 2015).
Key Event Description
DNA lesions may result from the formation of DNA adducts (i.e., covalent modification of DNA by chemicals), or by the action of agents such as radiation that may produce strand breaks or modified nucleotides within the DNA molecule. These DNA lesions are repaired through several mechanistically distinct pathways that can be categorized as follows:
- Damage reversal acts to reverse the damage without breaking any bonds within the sugar phosphate backbone of the DNA. The most prominent enzymes associated with damage reversal are photolyases (Sancar, 2003) that can repair UV dimers in some organisms, and O6-alkylguanine-DNA alkyltransferase (AGT) (Pegg 2011) and oxidative demethylases (Sundheim et al., 2008), which can repair some types of alkylated bases.
- Excision repair involves the removal of a damaged nucleotide(s) through cleavage of the sugar phosphate backbone followed by re-synthesis of DNA within the resultant gap. Excision repair of DNA lesions can be mechanistically divided into:
a) Base excision repair (BER) (Dianov and Hübscher, 2013), in which the damaged base is removed by a damage-specific glycosylase prior to incision of the phosphodiester backbone at the resulting abasic site. This leads to an intermediate that contains a DNA strand break, whereby DNA ligase is then recruited to seal the ends of the DNA.
b) Nucleotide excision repair (NER) (Schärer, 2013), in which the DNA strand containing the damaged nucleotide is incised at sites several nucleotides 5’ and 3’ to the site of damage, and a polynucleotide containing the damaged nucleotide is removed prior to DNA resynthesis within the resultant gap and sealing of the ends by DNA ligase.
c) Mismatch repair (MMR) (Li et al., 2016) which does not act on DNA lesions but does recognize mispaired bases resulting from replication errors. In MMR the strand containing the misincorporated base is removed prior to DNA resynthesis.
The major pathway that removes oxidative DNA damage is base excision repair (BER), which can be either monofunctional or bifunctional; in mammals, a specific DNA glycosylase (OGG1: 8-Oxoguanine glycosylase) is responsible for excision of 8-oxoguanine (8-oxoG) and other oxidative lesions (Hu et al., 2005; Scott et al., 2014; Whitaker et al., 2017). We note that long-patch BER is used for the repair of clustered oxidative lesions, which uses several enzymes from DNA replication pathways (Klungland and Lindahl, 1997). These pathways are described in detail in various reviews e.g., (Whitaker et al., 2017).
- Single strand break repair (SSBR) involves different proteins and enzymes depending on the origin of the SSB (e.g., produced as an intermediate in excision repair or due to direct chemical insult) but the same general steps of repair are taken for all SSBs: detection, DNA end processing, synthesis, and ligation (Caldecott, 2014). Poly-ADP-ribose polymerase1 (PARP1) detects and binds unscheduled SSBs (i.e., not deliberately induced during excision repair) and synthesizes PAR as a signal to the downstream factors in repair. PARP1 is not required to initiate SSBR of BER intermediates. The XRCC1 protein complex is then recruited to the site of damage where a common DNA intermediate as BER was generated, and acts as a scaffold for proteins and enzymes required for repair. Depending on the nature of the damaged termini of the DNA strand, different enzymes are required for end processing to generate the substrates that DNA polymerase β (Polβ; short patch repair) or Pol δ/ε (long patch repair) can bind to synthesize over the gap, although end processing is generally done by polynucleotide kinase. Synthesis in long-patch repair displaces a single stranded flap which is excised by flap endonuclease 1 (FEN1). In short-patch repair, the XRCC1/Lig3α complex joins the two ends after synthesis. In long-patch repair, the PCNA/Lig1 complex ligates the ends. (Caldecott, 2014).
- Double strand break repair (DSBR) is necessary to preserve genomic integrity when breaks occur in both strands of a DNA molecule. There are two major pathways for DSBR: homologous recombination (HR), which operates primarily during the S phase of dividing cell types, and nonhomologous end joining (NHEJ), which can function in both dividing and non-dividing cell types. No repair occurs in the M phase (Teruaki Iyama and David M. Wilson III, 2013). DNA repair in mitosis is controversial (Mladenov et al., 2023).
Complex lesions can be created by a single mutagen and can be more difficult to repair, as the mechanism behind how different repair pathways cooperate to address this is still unclear (Aleksandrov et al., 2018). In higher eukaryotes such as mammals, NHEJ is usually the preferred pathway for DNA DSBR. Its use, however, is dependent on the cell type, the gene locus, and the nuclease platform (Miyaoka et al., 2016). The use of NHEJ is also dependent on the cell cycle; NHEJ is generally not the pathway of choice when the cell is in the late S or G2 phase of the cell cycle, or in mitotic cells when the sister chromatid is directly adjacent to the double-strand break (DSB) (Lieber et al., 2003). In these cases, the HR pathway is commonly used for repair of DSBs. Despite this, NHEJ is still used more commonly than HR in human cells. Classical NHEJ (C-NHEJ) is the most common NHEJ repair mechanism, but alternative NHEJ (alt-NHEJ) can also occur, especially in the absence of C-NHEJ and HR.
The process of C-NHEJ in humans requires at least seven core proteins: Ku70, Ku86, DNA-dependent protein kinase complex (DNA-PKcs ), Artemis, X-ray cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF), and DNA ligase IV (Boboila et al., 2012). When DSBs occur, the Ku proteins, which have a high affinity for DNA ends, will bind to the break site and form a heterodimer. This protects the DNA from exonucleolytic attack and acts to recruit DNA-PKcs, the catalytic subunit, thus forming a trimeric complex on the ends of the DNA strands. Alternative NHEJ, or alt NHEJ, uses small similar sequences in two broken DNA ends to join them together. Unlike the usual repair method (cNHEJ), aNHEJ doesn't need specific proteins like LIG4 and KU. Instead, it relies on the MRN complex to process the breaks. However, alt NHEJ tends to cause mutations by adding or removing bits of DNA during the repair (Chaudhuri and Nussenzweig, 2017). The kinase activity of DNA-PKcs is then triggered, causing DNA-PKcs to auto-phosphorylate and thereby lose its kinase activity; the now phosphorylated DNA-PKcs dissociates from the DNA-bound Ku proteins. The free DNA-PKcs phosphorylates Artemis, an enzyme that possesses 5’-3’ exonuclease and endonuclease activity in the presence of DNA-PKcs and ATP. Artemis is responsible for ‘cleaning up’ the ends of the DNA. For 5’ overhangs, Artemis nicks the overhang, generally leaving a blunt duplex end. For 3’ overhangs, Artemis will often leave a four- or five-nucleotide single stranded overhang (Pardo et al., 2009; Fattah et al., 2010; Lieber et al., 2010). Next, the XLF and XRCC4 proteins form a complex which makes a channel to bind DNA and aligns the ends for efficient ligation via DNA ligase IV (Hammel et al., 2011).
The process of alt-NHEJ is less well understood than C-NHEJ and is a lower fidelity mechanism. Alt-NHEJ is known to involve slightly different core proteins than C-NHEJ and required microhomology repeats, but the steps of the pathway are essentially the same between the two processes (reviewed in Chiruvella et al., 2013). It is established, however, that alt-NHEJ is more error-prone in nature than C-NHEJ, which contributes to incorrect DNA repair. Alt-NHEJ is thus considered primarily to be a backup repair mechanism (reviewed in Chiruvella et al., 2013).
In contrast to NHEJ, HR takes advantage of similar or identical DNA sequences to repair DSBs and is not error-prone (Sung and Klein, 2006). The initiating step of HR is the creation of a 3’ single strand DNA (ss-DNA) overhang. Combinases such as RecA and Rad51 then bind to the ss-DNA overhang, and other accessory factors, including Rad54, help recognize and invade the homologous region on another DNA strand. From there, DNA polymerases are able to elongate the 3’ invading single strand and resynthesize the broken DNA strand using the corresponding sequence on the homologous strand.
Fidelity of DNA Repair
Most DNA repair pathways are extremely efficient. However, in principal, all DNA repair pathways can be overwhelmed when the DNA lesion burden exceeds the capacity of a given DNA repair pathway to recognize and remove the lesion. Exceeded repair capacity may lead to toxicity or mutagenesis following DNA damage. Apart from extremely high DNA lesion burden, inadequate repair may arise through several different specific mechanisms. For example, during repair of DNA containing O6-alkylguanine adducts, AGT irreversibly binds a single O6-alkylguanine lesion and as a result is inactivated (this is termed suicide inactivation, as its own action causes it to become inactivated). Thus, the capacity of AGT to carry out alkylation repair can become rapidly saturated when the DNA repair rate exceeds the de novo synthesis of AGT (Pegg, 2011).
A second mechanism relates to cell specific differences in the cellular levels or activity of some DNA repair proteins. For example, XPA is an essential component of the NER complex. The level of XPA that is active in NER is low in the testes, which may reduce the efficiency of NER in testes as compared to other tissues (Köberle et al., 1999). Likewise, both NER and BER have been reported to be deficient in cells lacking functional p53 (Adimoolam and Ford, 2003; Hanawalt et al., 2003; Seo and Jung, 2004). A third mechanism relates to the importance of the DNA sequence context of a lesion in its recognition by DNA repair enzymes. For example, 8-oxoguanine (8-oxoG) is repaired primarily by BER; the lesion is initially acted upon by a bifunctional glycosylase, OGG1, which carries out the initial damage recognition and excision steps of 8-oxoG repair. However, the rate of excision of 8-oxoG is modulated strongly by both chromatin components (Menoni et al., 2012) and DNA sequence context (Allgayer et al., 2013) leading to significant differences in the repair of lesions situated in different chromosomal locations.
DNA repair is also remarkably error-free. However, misrepair can arise during repair under some circumstances. DSBR is notably error prone, particularly when breaks are processed through NHEJ, during which partial loss of genome information is common at the site of the double strand break (Iyama and Wilson, 2013). This is because NHEJ rejoins broken DNA ends without the use of extensive homology; instead, it uses the microhomology present between the two ends of the DNA strand break to ligate the strand back into one. When the overhangs are not compatible, however, indels (insertion or deletion events), duplications, translocations, and inversions in the DNA can occur. These changes in the DNA may lead to significant issues within the cell, including alterations in the gene determinants for cellular fatality (Moore et al., 1996).
Activation of mutagenic DNA repair pathways to withstand cellular or replication stress either from endogenous or exogenous sources can promote cellular viability, albeit at a cost of increased genome instability and mutagenesis (Fitzgerald et al., 2017). These salvage DNA repair pathways including, Break-induced Replication (BIR) and Microhomology-mediated Break-induced Replication (MMBIR). BIR repairs one-ended DSBs and has been extensively studied in yeast as well as in mammalian systems. BIR and MMBIR are linked with heightened levels of mutagenesis, chromosomal rearrangements and ensuing genome instability (Deem et al., 2011; Sakofsky et al., 2015; Saini et al., 2017; Kramara et al., 2018). In mammalian genomes BIR-like synthesis has been proposed to be involved in late stage Mitotic DNA Synthesis (MiDAS) that predominantly occurs at so-called Common Fragile Sites (CFSs) and maintains telomere length under s conditions of replication stress that serve to promote cell viability (Minocherhomji et al., 2015; Bhowmick et al., 2016; Dilley et al., 2016).
Misrepair may also occur through other repair pathways. Excision repair pathways require the resynthesis of DNA and rare DNA polymerase errors during gap resynthesis will result in mutations (Brown et al., 2011). Errors may also arise during gap resynthesis when the strand that is being used as a template for DNA synthesis contains DNA lesions (Kozmin and Jinks-Robertson, 2013). In addition, it has been shown that sequences that contain tandemly repeated sequences, such as CAG triplet repeats, are subject to expansion during gap resynthesis that occurs during BER of 8-oxoG damage (Liu et al., 2009).
How it is Measured or Detected
There is no test guideline for this event. The event is usually inferred from measuring the retention of DNA adducts or the creation of mutations as a measure of lack of repair or incorrect repair. These ‘indirect’ measures of its occurrence are crucial to determining the mechanisms of genotoxic chemicals and for regulatory applications (i.e., determining the best approach for deriving a point of departure). More recently, a fluorescence-based multiplex flow-cytometric host cell reactivation assay (FM-HCR) has been developed to directly measure the ability of human cells to repair plasmid reporters (Nagel et al., 2014).
Indirect Measurement
In somatic and spermatogenic cells, measurement of DNA repair is usually inferred by measuring DNA adduct formation/removal. Insufficient repair is inferred from the retention of adducts and from increasing adduct formation with dose. Insufficient DNA repair is also measured by the formation of increased numbers of mutations and alterations in mutation spectrum. The methods will be specific to the type of DNA adduct that is under study.
Some EXAMPLES are given below for alkylated DNA.
DOSE-RESPONSE CURVE FOR ALKYL ADDUCTS/MUTATIONS: It is important to consider that some adducts are not mutagenic at all because they are very effectively repaired. Others are effectively repaired, but if these repair processes become overwhelmed mutations begin to occur. The relationship (shape of dose-response curve) between exposure to mutagenic agents and mutations provide an indication of whether the removal of adducts occurs, and whether it is more efficient at low doses. Sub-linear dose-response curves (hockey stick or j-shape curves) for mutation induction indicates that adducts are not converted to mutations at low doses. This suggests the effective repair of adducts at low doses, followed by saturation of repair at higher doses (Clewell et al., 2019). Thus, measurement of a clear point of inflection in the dose-response curve for mutations suggests that repair does occur, at least to some extent, at low dosees but that reduced repair efficiency arises above the inflection point. A lack of increase in mutation frequencies (i.e., flat line for dose-response) for a compound showing a dose-dependent increase in adducts would imply that the adducts formed are either not mutagenic or are effectively repaired.
RETENTION OF ALKYL ADDUCTS: Alkylated DNA can be found in cells long after exposure has occurred. This indicates that repair has not effectively removed the adducts. For example, DNA adducts have been measured in hamster and rat spermatogonia several days following exposure to alkylating agents, indicating lack of repair (Seiler et al., 1997; Scherer et al., 1987).
MUTATION SPECTRUM: Shifts in mutation spectrum (i.e., the specific changes in the DNA sequence) following a chemical exposure (relative to non-exposed mutation spectrum) indicates that repair was not operating effectively to remove specific types of lesions. The shift in mutation spectrum is indicative of the types of DNA lesions (target nucleotides and DNA sequence context) that were not repaired. For example, if a greater proportion of mutations occur at guanine nucleotides in exposed cells, it can be assumed that the chemical causes DNA adducts on guanine that are not effectively repaired.
Direct Measurement
Nagel et al. (2014) we developed a fluorescence-based multiplex flow-cytometric host cell reactivation assay (FM-HCR) to measures the ability of human cells to repair plasmid reporters. These reporters contain different types and amounts of DNA damage and can be used to measure repair through by NER, MMR, BER, NHEJ, HR and MGMT.
Please refer to the table below for additional details and methodologies for detecting DNA damage and repair.
| Assay Name | References | Description | DNA Damage/Repair Being Measured | OECD Approved Assay |
| Dose-Response Curve for Alkyl Adducts/ Mutations |
Lutz 1991
Clewell 2016 |
Creation of a curve plotting the stressor dose and the abundance of adducts/mutations; Characteristics of the resulting curve can provide information on the efficiency of DNA repair |
Alkylation, oxidative damage, or DSBs |
N/A |
| Retention of Alkyl Adducts |
Seiler 1997
Scherer 1987 |
Examination of DNA for alkylation after exposure to an alkylating agent; Presence of alkylation suggests a lack of repair | Alkylation | N/A |
| Mutation Spectrum | Wyrick 2015 | Shifts in the mutation spectrum after exposure to a chemical/mutagen relative to an unexposed subject can provide an indication of DNA repair efficiency, and can inform as to the type of DNA lesions present |
Alkylation, oxidative damage, or DSBs |
N/A |
| DSB Repair Assay (Reporter constructs) | Mao et al., 2011 | Transfection of a GFP reporter construct (and DsRed control) where the GFP signal is only detected if the DSB is repaired; GFP signal is quantified using fluorescence microscopy or flow cytometry | DSBs | N/A |
| Primary Rat Hepatocyte DNA Repair Assay |
Jeffrey and Williams, 2000
Butterworth et al., 1987 |
Rat primary hepatocytes are cultured with a 3H-thymidine solution in order to measure DNA synthesis in response to a stressor in non-replicating cells; Autoradiography is used to measure the amount of 3H incorporated in the DNA post-repair | Unscheduled DNA synthesis in response to DNA damage | N/A |
| Repair synthesis measurement by 3H-thymine incorporation | Iyama and Wilson, 2013 | Measure DNA synthesis in non-dividing cells as indication of gap filling during excision repair | Excision repair | N/A |
| Comet Assay with Time-Course |
Olive et al., 1990
Trucco et al., 1998 - Dunkenberger et al., 2022 |
Comet assay is performed with a time-course under alkaline conditions to detect SSBs and DSBs. Quantity of DNA in the tail should decrease as DNA repair progresses | DSBs | Yes (No. 489) |
| Flow Cytometry | Corneo et al., 2007 | The alt-NHEJ flow cytometer method involves utilizing an extrachromosomal substrate. Green fluorescent protein (GFP) expression is indicative of successful alt-NHEJ activity, contingent on the removal of 10 nucleotides from each end of the DNA and subsequent rejoining within a 9-nucleotide microhomology region. This approach provides a quantitative and visual means to measure the efficiency of alternative non-homologous end joining in cellular processes. | Alt NHEJ | No |
| Pulsed Field Gel Electro-phoresis (PFGE) with Time-Course | Biedermann et al., 1991 | PFGE assay with a time-course; Quantity of small DNA fragments should decrease as DNA repair progresses | DSBs | N/A |
|
Fluorescence -Based Multiplex Flow-Cytometric Host Reactivation Assay (FM-HCR) |
Nagel et al., 2014 | Measures the ability of human cells to repair plasma reporters, which contain different types and amounts of DNA damage; Used to measure repair processes including HR, NHEJ, BER, NER, MMR, and MGMT | HR, NHEJ, BER, NER, MMR, or MGMT | N/A |
| Alkaline Unwinding Assay with Time Course | Nacci et al. 1991 | DNA is stored in alkaline solutions with DNA-specific dye and allowed to unwind following removal from tissue, increased strand damage associated with increased unwinding. Samples analyzed at different time points to compare remaining damage following repair opportunities | DSBs | Yes (No. 489) |
| Sucrose Density Gradient Centrifugation with Time Course | Larsen et al. 1982 | Strand breaks alter the molecular weight of the DNA piece. DNA in alkaline solution centrifuged into sugar density gradient, repeated set time apart. The less DNA breaks identified in the assay repeats, the more repair occurred | SSBs | N/A |
| y-H2AX Foci Staining with Time Course |
Mariotti et al. 2013 Penninckx et al. 2021 |
Histone H2AX is phosphorylated in the presence of DNA strand breaks, the rate of its disappearance over time is used as a measure of DNA repair | DSBs | N/A |
| Alkaline Elution Assay with Time Course | Larsen et al. 1982 | DNA with strand breaks elute faster than DNA without, plotted against time intervals to determine the rate at which strand breaks repair | SSBs | N/A |
| 53BP1 foci Detection with Time Course | Penninckx et al. 2021 | 53BP1 is recruited to the site of DNA damage, the rate at which its level decreases over time is used to measure DNA repair | DSBs | N/A |
References
Adimoolam, S. & J.M. Ford (2003), "p53 and regulation of DNA damage recognition during nucleotide excision repair" DNA Repair (Amst), 2(9): 947-54.
Aleksandrov, Radoslav et al. (2018), “Protein Dynamics in Complex DNA Lesions.” Molecular cell,69(6): 1046-1061.e5. doi:10.1016/j.molcel.2018.02.016
Allgayer, J. et al. (2013), "Modulation of base excision repair of 8-oxoguanine by the nucleotide sequence", Nucleic Acids Res, 41(18): 8559-8571. Doi: 10.1093/nar/gkt620.
Beranek, D.T. (1990), "Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents", Mutation Research, 231(1): 11-30. Doi: 10.1016/0027-5107(90)90173-2.
Bhatti, A. et al., (2016), “Homologous Recombination Biology.”, Encyclopedia Britannica.
Bhowmick, R., S. et al. (2016), "RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress", Mol Cell, 64:1117-1126. Doi: 10.1016/j.molcel.2016.10.037.
Biedermann, A. K. et al. (1991), “SCID mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair”, Cell Biology, 88(4): 1394-7. Doi: 10.1073/pnas.88.4.1394.
Boboila, C., F. W. Alt & B. Schwer. (2012), “Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks.” Adv Immunol, 116, 1-49. doi:10.1016/B978-0-12-394300-2.00001-6
Bronstein, S.M. et al. (1991), "Toxicity, mutagenicity, and mutational spectra of N-ethyl-N-nitrosourea in human cell lines with different DNA repair phenotypes", Cancer Research, 51(19): 5188-5197.
Bronstein, S.M. et al. (1992), "Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells", Cancer Research, 52(7): 2008-2011.
Brown, J.A. et al. (2011), "Efficiency and fidelity of human DNA polymerases λ and β during gap-filling DNA synthesis", DNA Repair (Amst)., 10(1):24-33.
Butterworth, E. B. et al., (1987), A protocol and guide for the in vitro rat hepatocyte DNA-repair assay. Mutation Research. 189, 113-21. Doi: 10.1016/0165-1218(87)90017-6.
Caldecott, K. W. (2014), "DNA single-strand break repair", Exp Cell Res, 329(1): 2-8.
Chaudhuri, R.A. and Nussenzweig, A. (2017), “The multifaceted roles of PARP1 in DNA repair and chromatin remodelling”. Nat Rev Mol Cell Biol 18, 610–621. https://doi.org/10.1038/nrm.2017.53
Chen, L. et al., (2001), Promotion of DNA ligase IV-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell. 8(5), 1105-15.
Chiruvella, K. K., Z. Liang & T. E. Wilson, (2013), Repair of Double-Strand Breaks by End Joining. Cold Spring Harbor Perspectives in Biology, 5(5):127-57. Doi: 10.1101/cshperspect.a012757.
Clewell, R. A. et al. (2019). “Dose-dependence of chemical carcinogenicity: Biological mechanisms for thresholds and implications for risk assessment”. Chem Biol Interact. 2019 Mar 1;301:112-127. doi: 10.1016/j.cbi.2019.01.025.
Corneo, B. et al., 2007, "Rag mutations reveal robust alternative end joining”. Nature 449, 483–486 (2007). https://doi.org/10.1038/nature06168
Dahle, J., et al. (2008), “Overexpression of human OGG1 in mammalian cells decreases ultraviolet A induced mutagenesis”, Cancer Letters, Vol.267, Elsevier, Amsterdam, https://doi.org/10.1016/j.canlet.2008.03.002.
Deem, A. et al. (2011), "Break-Induced Replication Is Highly Inaccurate.", PLoS Biol. 9:e1000594. Doi: 10.1371/journal.pbio.1000594.
Dianov, G.L. & U. Hübscher (2013), "Mammalian base excision repair: the forgotten archangel", Nucleic Acids Res., 41(6):3483-90. Doi: 10.1093/nar/gkt076.
Dilley, R.L. et al. Greenberg (2016), "Break-induced telomere synthesis underlies alternative telomere maintenance", Nature, 539:54-58. Doi: 10.1038/nature20099.
Douglas, G.R. et al. (1995), "Temporal and molecular characteristics of mutations induced by ethylnitrosourea in germ cells isolated from seminiferous tubules and in spermatozoa of lacZ transgenic mice", Proceedings of the National Academy of Sciences of the United States of America, 92(16):7485-7489. Doi: 10.1073/pnas.92.16.7485.
Dunkenberger, Logan et al. (2022), “Comet Assay for the Detection of Single and Double-Strand DNA Breaks.” Methods in molecular biology (Clifton, N.J.), 2422: 263-269. doi:10.1007/978-1-0716-1948-3_18
Fattah, F. et al., (2010), Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet, 6(2), doi:10.1371/journal.pgen.1000855
Fitzgerald, D.M., P.J. Hastings, and S.M. Rosenberg (2017), "Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance", Ann Rev Cancer Biol, 1:119-140. Doi: 10.1146/annurev-cancerbio-050216-121919.
Gorbunova, V. and A. Seluanov. (2016), “DNA double strand break repair, aging and the chromatin connection”, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Vol.788/1-2, Elsevier, Amsterdam, http://dx.doi.org/10.1016/j.mrfmmm.2016.02.004.
Hammel, M. et al., (2011), XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem, 286(37), 32638-32650. doi:10.1074/jbc.M111.272641.
Hanawalt, P.C., J.M. Ford and D.R. Lloyd (2003), "Functional characterization of global genomic DNA repair and its implications for cancer", Mutation Research, 544(2-3): 107–114.
Harbach, P. R. et al., (1989), “The in vitro unscheduled DNA synthesis (UDS) assay in rat primary hepatocytes”, Mutation Research, 216(2):101-10. Doi:10.1016/0165-1161(89)90010-1.
Iyama, T. and D.M. Wilson III (2013), "DNA repair mechanisms in dividing and non-dividing cells", DNA Repair, 12(8): 620– 636.
Jeffrey, M. A.& M. G. Williams, (2000), “Lack of DNA-damaging Activity of Five Non-nutritive Sweeteners in the Rat Hepatocyte/DNA Repair Assay”, Food and Chemical Toxicology, 38: 335-338. Doi: 10.1016/S0278-6915(99)00163-5.
Köberle, B. et al. (1999), "Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours", Curr. Biol., 9(5):273-6. Doi: 10.1016/s0960-9822(99)80118-3.
Kozmin, S.G. & S. Jinks-Robertson S. (2013), “The mechanism of nucleotide excision repair-mediated UV-induced mutagenesis in nonproliferating cells”, Genetics, 193(3): 803-17. Doi: 10.1534/genetics.112.147421.
Kramara, J., B. Osia, and A. Malkova (2018), "Break-Induced Replication: The Where, The Why, and The How", Trends Genet, 34:518-531. Doi: 10.1016/j.tig.2018.04.002.
Kuhne, M., G. Urban and M. Lo (2005), "DNA Double-Strand Break Misrejoining after Exposure of Primary Human Fibroblasts to CK Characteristic X Rays, 29 kVp X Rays and 60Co γ Rays", Radiation. Research, Vol.164/5, Radiation Research Society, Indianapolis, https://doi.org/10.1667/RR3461.1.
Larsen, K.H. et al. (1982), “DNA repair assays as tests for environmental mutagens: A report of the U.S. EPA gene-tox program”, Mutation Research, Vol.98/3, Elsevier, Amsterdam, https://doi.org/10.1016/0165-1110(82)90037-9.
Li Z, A. H. Pearlman, and P. Hsieh (2016), "DNA mismatch repair and the DNA damage response", DNA Repair (Amst), 38:94-101.
Lieber, M. R., (2010), “The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway.” Annu Rev Biochem. 79:181-211. doi:10.1146/annurev.biochem.052308.093131.
Lieber, M. R. et al., (2003), “Mechanism and regulation of human non-homologous DNA end-joining”, Nat Rev Mol Cell Biol. 4(9):712-720. doi:10.1038/nrm1202.
Liu, Y. et al. (2009), "Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion", J. Biol. Chem., 284(41): 28352-28366. Doi: 10.1074/jbc.M109.050286.
Mao, Z. et al., (2011), “SIRT6 promotes DNA repair under stress by activating PARP1”, Science. 332(6036): 1443-1446. doi:10.1126/science.1202723.
Mariotti, L.G. et al. (2013), “Use of the γ-H2AX Assay to Investigate DNA Repair Dynamics Following Multiple Radiation Exposures”, PLoS ONE, Vol.8/11, PLoS, San Francisco, https://doi.org/10.1371/journal.pone.0079541.
Matthews, L. A., & L. A. Simmons, (2014), “Bacterial nonhomologous end joining requires teamwork”, J Bacteriol. 196(19): 3363-3365. doi:10.1128/JB.02042-14.
Menoni, H. et al. (2012), "Base excision repair of 8-oxoG in dinucleosomes", Nucleic Acids Res. ,40(2): 692-700. Doi: 10.1093/nar/gkr761.
Minocherhomji, S. et al. (2015), "Replication stress activates DNA repair synthesis in mitosis", Nature, 528:286-290. Doi: 10.1038/nature16139.
Miyaoka, Y. et al., (2016), “Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing”, Sci Rep, 6, 23549. doi:10.1038/srep23549/.
Mladenov. et al. (2023), . “New Facets of DNA Double Strand Break Repair: Radiation Dose as Key Determinant of HR versus c-NHEJ Engagement”. International journal of molecular sciences, 24(19), 14956. https://doi.org/10.3390/ijms241914956
Moore, J. K., & J. E. Haber, (1996), “Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae”, Molecular and Cellular Biology, 16(5), 2164–73. Doi: 10.1128/MCB.16.5.2164.
Nacci, D. et al. (1992), “Application of the DNA alkaline unwinding assay to detect DNA strand breaks in marine bivalves”, Marine Environmental Research, Vol.33/2, Elsevier BV, Amsterdam, https://doi.org/10.1016/0141-1136(92)90134-8.
Nagel, Z.D. et al. (2014), "Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis", Proc. Natl. Acad. Sci. USA, 111(18):E1823-32. Doi: 10.1073/pnas.1401182111.
O’Brien, J.M. et al. (2015), "Sublinear response in lacZ mutant frequency of Muta™ Mouse spermatogonial stem cells after low dose subchronic exposure to N-ethyl-N-nitrosourea", Environ. Mol. Mutagen., 56(4): 347-55. Doi: 10.1002/em.21932.
Olive, L. P., J. P. Bnath & E. R. Durand, (1990), “Heterogeneity in Radiation-Induced DNA Damage and Repairing Tumor and Normal Cells Measured Using the "Comet" Assay”, Radiation Research. 122: 86-94. Doi: 10.1667/rrav04.1.
Pardo, B., B. Gomez-Gonzalez & A. Aguilera, (2009), “DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship”, Cell Mol Life Sci, 66(6), 1039-1056. doi:10.1007/s00018-009-8740-3.
Pegg, A.E. (2011), "Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools", Chem. Res. Toxicol., 4(5): 618-39. Doi: 10.1021/tx200031q.
Penninckx, S. et al. (2021), “Quantification of radiation-induced DNA double strand break repair foci to evaluate and predict biological responses to ionizing radiation”, NAR Cancer, Vol.3/4, Oxford University Press, Oxford, https://doi.org/10.1093/narcan/zcab046.
Rydberg, B. et al. (2005), "Dose-Dependent Misrejoining of Radiation-Induced DNA Double-Strand Breaks in Human Fibroblasts: Experimental and Theoretical Study for High- and Low-LET Radiation", Radiation Research, Vol.163/5, Radiation Research Society, Indianapolis, https://doi.org/10.1667/RR3346.
Sancar, A. (2003), "Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors", Chem Rev., 103(6): 2203-37. Doi: 10.1021/cr0204348.
Saini, N. et al. (2017), "Migrating bubble during break-induced replication drives conservative DNA synthesis", Nature, 502:389-392. Doi: 10.1038/nature12584.
Sakofsky, C.J. et al. (2015), "Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements", Mol Cell, 60:860-872. Doi: 10.1016/j.molcel.2015.10.041.
Schärer, O.D. (2013), "Nucleotide excision repair in eukaryotes", Cold Spring Harb. Perspect. Biol., 5(10): a012609. Doi: 10.1101/cshperspect.a012609.
Scherer, E., A.A. Jenner and L. den Engelse (1987), "Immunocytochemical studies on the formation and repair of O6-alkylguanine in rat tissues", IARC Sci Publ., 84: 55-8.
Seiler, F., K. Kamino, M. Emura, U. Mohr and J. Thomale (1997), "Formation and persistence of the miscoding DNA alkylation product O6-ethylguanine in male germ cells of the hamster", Mutat Res., 385(3): 205-211. Doi: 10.1016/s0921-8777(97)00043-8.
Shelby, M.D. and K.R. Tindall (1997), "Mammalian germ cell mutagenicity of ENU, IPMS and MMS, chemicals selected for a transgenic mouse collaborative study", Mutation Research, 388(2-3): 99-109. Doi: 10.1016/s1383-5718(96)00106-4.
Seo, Y.R. and H.J. Jung (2004), "The potential roles of p53 tumor suppressor in nucleotide excision repair (NER) and base excision repair (BER)", Exp. Mol. Med., 36(6): 505-509. Doi: 10.1038/emm.2004.64.
Sundheim, O. et al. (2008), "AlkB demethylases flip out in different ways", DNA Repair (Amst)., 7(11): 1916-1923. Doi: 10.1016/j.dnarep.2008.07.015.
Sung, P., & H. Klein, (2006), “Mechanism of homologous recombination: mediators and helicases take on regulatory functions”, Nat Rev Mol Cell Biol, 7(10), 739-750. Doi:10. 1038/nrm2008.
Trucco, C., et al., (1998), “DNA repair defect i poly(ADP-ribose) polymerase-deficient cell lines”, Nucleic Acids Research. 26(11): 2644–2649. Doi: 10.1093/nar/26.11.2644.
Trzeciak, A.R. et al. (2008), “Age, sex, and race influence single-strand break repair capacity in a human population”, Free Radical Biology & Medicine, Vol. 45, Elsevier, Amsterdam, https://doi.org/10.1016/j.freeradbiomed.2008.08.031.
White, R.R. and J. Vijg. (2016), “Do DNA Double-Strand Breaks Drive Aging?”, Molecular Cell, Vol.63, Elsevier, Amsterdam, http://doi.org/10.1016/j.molcel.2016.08.004.
Wyrick, J.J. & S. A. Roberts, (2015), “Genomic approaches to DNA repair and mutagenesis”, DNA Repair (Amst). 36:146-155. doi: 10.1016/j.dnarep.2015.09.018.
van Zeeland, A.A., A. de Groot and A. Neuhäuser-Klaus (1990), "DNA adduct formation in mouse testis by ethylating agents: a comparison with germ-cell mutagenesis", Mutat. Res., 231(1): 55-62.
Event: 1635: Increase, DNA strand breaks
Short Name: Increase, DNA strand breaks
Event Component
| Process | Object | Action |
|---|---|---|
| DNA Strand Break | Deoxyribonucleic acid | increased |
AOPs Including This Key Event
Stressors
| Name |
|---|
| Ionizing Radiation |
| Topoisomerase inhibitors |
| Radiomimetic compounds |
Biological Context
| Level of Biological Organization |
|---|
| Molecular |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human and other cells in culture | human and other cells in culture | NCBI |
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
Taxonomic applicability: DNA strand breaks are relevant to all species, including vertebrates such as humans, that contain DNA (Cannan & Pederson, 2016).
Life stage applicability: This key event is not life stage specific as all life stages display strand breaks. However, there is an increase in baseline levels of DNA strand breaks seen in older individuals though it is unknown whether this change due to increased break induction or a greater retention of breaks due to poor repair (White & Vijg, 2016).
Sex applicability: This key event is not sex specific as both sexes display evidence of strand breaks. In some cell types, such as peripheral blood mononuclear cells, males show higher levels of single strand breaks than females (Garm et al., 2012).
Evidence for perturbation by a stressor: There are studies demonstrating that increased DNA strand breaks can result from exposure to multiple stressor types including ionizing & non-ionizing radiation, chemical agents, and oxidizing agents (EPRI, 2014; Hamada, 2014; Cencer et al., 2018; Cannan & Pederson, 2016; Yang et al., 1998).
Key Event Description
DNA strand breaks are a type of damage resulting from the hydrolysis of phosphodiester groups in the backbone of DNA molecules (Gates, 2009) and can occur on a single strand (single strand breaks; SSBs) or both strands (double strand breaks; DSBs). SSBs arise when the sugar phosphate backbones connecting adjacent nucleotides in DNA are simultaneously hydrolyzed such that the hydrogen bonds between complementary bases are not able to hold the two strands together. DSBs are generated when both strands are simultaneously broken at sites that are sufficiently close to one another that base-pairing and chromatin structure are insufficient to keep the two DNA ends juxtaposed. As a consequence, the two DNA ends generated by a DSB can physically dissociate from one another, becoming difficult to repair and increasing the chance of inappropriate recombination with other sites in the genome (Jackson, 2002). SSB can turn into DSB if the replication fork stalls at the lesion leading to fork collapse. Strand breaks are intermediates in various biological events, including DNA repair (e.g., excision repair), as well as other normal cellular processes where DSBs act as genetic shufflers to generate genetic diversity for V(D)J recombination in lymphoid cells, and chromatin remodeling in both somatic cells and germ cells, and meiotic recombination in gametes.
Strand breaks are intermediates in various biological events, including DNA repair (e.g., excision repair), V(D)J recombination in developing lymphoid cells and chromatin remodeling in both somatic cells and germ cells. The spectrum of damage can be complex, particularily if the stressor is from large amounts of deposited energy which can result in complex lesions and clustered damage defined as two or more oxidized bases, abasic sites or strand breaks on opposing DNA strands within a few helical turns. These lesions are more difficult to repair and have been studied in many types of models (Barbieri et al., 2019 and Asaithamby et al., 2011). DSBs and complex lesions are of particular concern, as they are considered the most lethal and deleterious type of DNA lesion. If misrepaired or left unrepaired, DSBs may drive the cell towards genomic instability, apoptosis or tumorigenesis (Beir, 1999).
How it is Measured or Detected
Please refer to the table below for details regarding these and other methodologies for detecting DNA DSBs.
|
Method of Measurement |
References |
Description |
OECD Approved Method? |
|
Comet Assay (Single Cell Gel Eletrophoresis - Alkaline) |
Collins, 2004; Olive and Banath, 2006; Platel et al., 2011; Nikolova et al., 2017 |
To detect SSBs or DSBs, single cells are encapsulated in agarose on a slide, lysed, and subjected to gel electrophoresis at an alkaline pH (pH >13); DNA fragments are forced to move, forming a "comet"-like appearance |
Yes |
|
γ-H2AX Foci Quantification - Flow Cytometry |
Rothkamm and Horn, 2009; Bryce et al., 2016 |
Measurement of γ-H2AX immunostaining in cells by flow cytometry, normalized to total levels of H2AX |
No |
|
γ-H2AX Foci Quantification - Western Blot |
Burma et al., 2001; Revet et al., 2011 |
Measurement of γ-H2AX immunostaining in cells by Western blotting, normalized to total levels of H2AX |
No |
|
γ-H2AX Foci Quantification - Microscopy |
Redon et al., 2010; Mah et al., 2010; Garcia-Canton et al., 2013 |
Quantification of γ-H2AX immunostaining by counting γ-H2AX foci visualized with a microscope |
No |
|
γ-H2AX Foci Quantification - ELISA |
Ji et al., 2017 |
Measurement of γ-H2AX in cells by ELISA, normalized to total levels of H2AX |
No |
|
Pulsed Field Gel Electrophoresis (PFGE) |
Ager et al., 1990; Gardiner et al., 1985; Herschleb et al., 2007; Kawashima et al., 2017 |
To detect DSBs, cells are embedded and lysed in agarose, and the released DNA undergoes gel electrophoresis in which the direction of the voltage is periodically alternated; Large DNA fragments are thus able to be separated by size |
No |
|
The TUNEL (Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling) Assay |
Loo, 2011 |
To detect strand breaks, dUTPs added to the 3’OH end of a strand break by the DNA polymerase terminal deoxynucleotidyl transferase (TdT) are tagged with a fluorescent dye or a reporter enzyme to allow visualization |
No |
|
In Vitro DNA Cleavage Assays using Topoisomerase |
Nitiss, 2012 |
Cleavage of DNA can be achieved using purified topoisomerase; DNA strand breaks can then be separated and quantified using gel electrophoresis |
No |
|
PCR assay |
Figueroa‑González & Pérez‑Plasencia, 2017 |
Assay of strand breaks through the observation of DNA amplification prevention. Breaks block Taq polymerase, reducing the number of DNA templates, preventing amplification |
No |
|
Sucrose density gradient centrifuge |
Raschke et al. 2009 |
Division of DNA pieces by density, increased fractionation leads to lower density pieces, with the use of a sucrose cushion |
No |
|
Alkaline Elution Assay |
Kohn, 1991 |
Cells lysed with detergent-solution, filtered through membrane to remove all but intact DNA |
No |
|
Unwinding Assay |
Nacci et al. 1992 |
DNA is stored in alkaline solutions with DNA-specific dye and allowed to unwind following removal from tissue, increased strand damage associated with increased unwinding |
Yes |
|
STRIDE assay |
Zilio and Ulrich, 2021 |
STRIDE (SensiTive Recognition of Individual DNA Ends) combines in situ nick translation with the proximity ligation assay (PLA) to detect single-strand breaks (sSTRIDE) or double-strand breaks (dSTRIDE). In this process, lesions labeled through nick translation with biotinylated nucleotides are identified by a PLA signal, which arises from the interaction of two anti-biotin antibodies from different species.
|
No |
|
sBLISS |
Bouwmann et al. 2020 |
sBLISS (in-suspension breaks labeling in situ and sequencing) labels double-strand breaks (DSBs) in cells immobilized on glass coverslips, using double-stranded oligonucleotide adaptors that facilitate selective linear amplification through T7-mediated in vitro transcription (IVT), followed by next-generation sequencing (NGS) library preparation
|
No |
References
Ager, D. D., et al. (1990). Measurement of radiation-induced DNA double-strand breaks by pulsed-field gel electrophoresis. Radiation research, 122/(2), 181–187.
Anderson, D. & Laubenthal J. (2013), “Analysis of DNA Damage via Single-Cell Electrophoresis. In: Makovets S, editor. DNA Electrophoresis. Totowa.”, NJ: Humana Press. p 209-218.
Asaithamby, A., B. Hu and D.J. Chen. (2011) “Unrepaired clustered DNA lesions induce chromosome breakage in human cells.” Proc Natl Acad Sci U S A 108(20): 8293-8298 .
Barbieri, S., G. Babini, J. Morini et a l (2019). . Predicting DNA damage foci and their experimental readout with 2D microscopy: a unified approach applied to photon and neutron exposures. Scientific Reports 9(1): 14019
Bouwman, B. et al. (2020), “Genome-wide detection of DNA double-strand breaks by in-suspension BLISS”, Nature protocols,.15/12, Springer Nature, London, https://doi.org/10.1038/s41596-020-0397-2
Bryce, S. et al. (2016), “Genotoxic mode of action predictions from a multiplexed flow cytometric assay and a machine learning approach.”, Environ Mol Mutagen. 57:171-189. Doi: 10.1002/em.21996.
Burma, S. et al. (2001), “ATM phosphorylates histone H2AX in response to DNA double-strand breaks.”, J Biol Chem, 276(45): 42462-42467. doi:10.1074/jbc.C100466200
Cannan, W.J. and D.S. Pederson (2016), "Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin.", Journal of Cellular Physiology, Vol.231(/1), Wiley, New York, https://doi.org/10.1002/jcp.25048.
Cencer, C. et al. (2018), “PARP-1/PAR Activity in Cultured Human Lens Epithelial Cells Exposed to Two Levels of UVB Light”, Photochemistry and Photobiology, Vol.(94/1), Wiley-Blackwell, Hoboken, https://doi.org/10.1111/php.12814.
Charlton, E. D. et al. (1989), “Calculation of Initial Yields of Single and Double Stranded Breaks in Cell Nuclei from Electrons, Protons, and Alpha Particles.”, Int. J. Radiat. Biol. 56(1): 1-19. doi: 10.1080/09553008914551141.
Collins, R. A. (2004), “The Comet Assay for DNA Damage and Repair. Molecular Biotechnology.”, Mol Biotechnol. 26(3): 249-61. doi:10.1385/MB:26:3:249
EPRI (2014), Epidemiology and mechanistic effects of radiation on the lens of the eye: Review and scientific appraisal of the literature, EPRI, California.
Figueroa‑González, G. and C. Pérez‑Plasencia. (2017), “Strategies for the evaluation of DNA damage and repair mechanisms in cancer”, Oncology Letters, Vol.133(/6), Spandidos Publications, Athens, https://doi.org/10.3892/ol.2017.6002.
Garcia-Canton, C. et al. (2013), “Assessment of the in vitro p-H2AX assay by High Content Screening asa novel genotoxicity test.”, Mutat Res. 757:158-166. Doi: 10.1016/j.mrgentox.2013.08.002
Gardiner, K. et al. (1986), “Fractionation of Large Mammalian DNA Restriction Fragments Using Vertical Pulsed-Field Gradient Gel Electrophoresis.”, Somatic Cell and Molecular Genetics. 12(2): 185-95.Doi: 10.1007/bf01560665.
Garm, C. et al. (2012), “Age and gender effects on DNA strand break repair in peripheral blood mononuclear cells”, Aging Cell, Vol.12/1, Blackwell Publishing Ltd, Oxford, https://doi.org/10.1111/acel.12019.
Hamada, N. (2014), “What are the intracellular targets and intratissue target cells for radiation effects?”, Radiation research, Vol. 181/1, The Radiation Research Society, Indianapolis, https://doi.org/10.1667/RR13505.1.
Herschleb, J. et al. (2007), “Pulsed-field gel electrophoresis.”, Nat Protoc. 2(3): 677-684. doi:10.1038/nprot.2007.94
Iliakis, G. et al. (2015), “Alternative End-Joining Repair Pathways Are the Ultimate Backup for Abrogated Classical Non-Homologous End-Joining and Homologous Recombination Repair: Implications for the Formation of Chromosome Translocations.”, Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2(3): 677-84. doi: 10.1038/nprot.2007.94
Jackson, S. (2002). “Sensing and repairing DNA double-strand breaks.”, Carcinogenesis. 23:687-696. Doi:10.1093/carcin/23.5.687.
Ji, J. et al. (2017), “Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay.”, PLoS One. 12(2): e0171582. doi:10.1371/journal.pone.0171582
Kawashima, Y.(2017), “Detection of DNA double-strand breaks by pulsed-field gel electrophoresis.”, Genes Cells 22:84-93. Doi: 10.1111/gtc.12457.
Khoury, L. et al. (2013), “Validation of high-throughput genotoxicity assay screening using cH2AX in-cell Western assay on HepG2 cells.”, Environ Mol Mutagen, 54:737-746. Doi: 10.1002/em.21817.
Khoury, L. et al. (2016), “Evaluation of four human cell lines with distinct biotransformation properties for genotoxic screening.”, Mutagenesis, 31:83-96. Doi: 10.1093/mutage/gev058.
Kohn, K.W. (1991), “Principles and practice of DNA filter elution”, Pharmacology & Therapeutics, Vol.49(/1), Elsevier, Amsterdam, https://doi.org/10.1016/0163-7258(91)90022-E.
Loo, DT. (2011), “In Situ Detection of Apoptosis by the TUNEL Assay: An Overview of Techniques. In: Didenko V, editor. DNA Damage Detection In Situ, Ex Vivo, and In Vivo. Totowa.”, NJ: Humana Press. p 3-13.doi: 10.1007/978-1-60327-409-8_1.
Mah, L. J. et al. (2010), “Quantification of gammaH2AX foci in response to ionising radiation.”, J Vis Exp(38). doi:10.3791/1957.
Nacci, D. et al. (1992), “Application of the DNA alkaline unwinding assay to detect DNA strand breaks in marine bivalves”, Marine Environmental Research, Vol.33(/2), Elsevier BV, Amsterdam, https://doi.org/10.1016/0141-1136(92)90134-8.
Nikolova, T., F. et al. (2017), “Genotoxicity testing: Comparison of the γH2AX focus assay with the alkaline and neutral comet assays.”, Mutat Res 822:10-18. Doi: 10.1016/j.mrgentox.2017.07.004.
Nitiss, J. L. et al. (2012), “Topoisomerase assays. ”, Curr Protoc Pharmacol. Chapter 3: Unit 3 3.
OECD. (2014). Test No. 489: “In vivo mammalian alkaline comet assay.” OECD Guideline for the Testing of Chemicals, Section 4 .
Olive, P. L., & Banáth, J. P. (2006), “The comet assay: a method to measure DNA damage in individual cells.”, Nature Protocols. 1(1): 23-29. doi:10.1038/nprot.2006.5.
Platel A. et al. (2011), “Study of oxidative DNA damage in TK6 human lymphoblastoid cells by use of the thymidine kinase gene-mutation assay and the in vitro modified comet assay: Determination of No-Observed-Genotoxic-Effect-Levels.”, Mutat Res 726:151-159. Doi: 10.1016/j.mrgentox.2011.09.003.
Raschke, S., J. Guan and G. Iliakis. (2009), “Application of alkaline sucrose gradient centrifugation in the analysis of DNA replication after DNA damage”, Methods in Molecular Biology, Vol.521, Humana Press, Totowa, https://doi.org/10.1007/978-1-60327-815-7_18.
Redon, C. et al. (2010), “The use of gamma-H2AX as a biodosimeter for total-body radiation exposure in non-human primates.”, PLoS One. 5(11): e15544. doi:10.1371/journal.pone.0015544
Revet, I. et al. (2011), “Functional relevance of the histone γH2Ax in the response to DNA damaging agents.” Proc Natl Acad Sci USA.108:8663-8667. Doi: 10.1073/pnas.1105866108
Rogakou, E.P. et al. (1998), “DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139.” , J Biol Chem, 273:5858-5868. Doi: 10.1074/jbc.273.10.5858
Rothkamm, K. & Horn, S. (2009), “γ-H2AX as protein biomarker for radiation exposure.”, Ann Ist Super Sanità, 45(3): 265-71.
White, R.R. and J. Vijg. (2016), “Do DNA Double-Strand Breaks Drive Aging?”, Molecular Cell, Vol.63, Elsevier, Amsterdam, http://doi.org/10.1016/j.molcel.2016.08.004.
Yang, Y. et al. (1998), “The effect of catalase amplification on immortal lens epithelial cell lines”, Experimental Eye Research, Vol.67(/6), Academic Press Inc, Cambridge, https://doi.org/10.1006/exer.1998.0560.
Zilio, N. and H. D. Ulrich (2021), “Exploring the SSBreakome: genome-wide mapping of DNA single-strand breaks by next-generation sequencing”, The FEBS journal, 288(13), Wiley, Hoboken, https://doi.org/10.1111/febs.15568
Event: 1505: Increase, Cell cycle disruption
Short Name: Cell cycle disruption
Event Component
| Process | Object | Action |
|---|---|---|
| regulation of cell cycle | cell cycle-related cyclin | disrupted |
AOPs Including This Key Event
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| cell |
Organ term
| Organ term |
|---|
| organ |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Mus musculus | Mus musculus | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Not Otherwise Specified | Moderate |
| Sex | Evidence |
|---|---|
| Unspecific | High |
The histone gene expression alters in each phase of the cell cycle in human HeLa cells (Homo sapiens) [Heintz et al., 1982].
Key Event Description
The disruption of the cell cycle leads to a decrease in cell number. The cell cycle consists of G1, S, G2, M, and G0 phases. The cell cycle regulation is disrupted by the cell cycle arrest in certain cell cycle phases. The histone gene expression is regulated in cell cycle phases [Heintz et al., 1983].
How it is Measured or Detected
The percentage of cells at G1, G0, S, and G2/M phases can be detected by flow cytometry [Li et al., 2013]. Cell cycle distribution was analyzed by fluorescence-activated cell sorter (FACS) analysis with a Partec PAS-II sorter [Zupkovitz et al., 2010]. The four cell-cycle phases in living cells can be measured with four-color fluorescent proteins using live-cell imaging [Bajar et al., 2016]. The incorporation of [3H]deoxycytidine or [3H]thymidine into cell DNA during the S phase can be monitored as DNA synthesis [Heintz et al., 1982].
References
Bajar, B.T. et al. (2016), "Fluorescent indicators for simultaneous reporting of all four cell cycle phases", Nat Methods 13:993-996
Heintz, N. et al. (1983), "Regulation of human histone gene expression: Kinetics of accumulation and changes in the rate of synthesis and in the half-lives of individual histone mRNAs during the HeLa cell cycle", Molecular and Cellular Biology 3:539-550
Li, Q. et al. (2013), "Glyphosate and AMPA inhibit cancer cell growth through inhibiting intracellular glycine synthesis", Drug Des Devel Ther 7:635-643
Event: 1821: Decrease, Cell proliferation
Short Name: Decrease, Cell proliferation
Event Component
| Process | Object | Action |
|---|---|---|
| cell proliferation | cell | decreased |
AOPs Including This Key Event
Stressors
| Name |
|---|
| 2,4-Dinitrophenol |
| Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
| Carbonyl cyanide m-chlorophenyl hydrazone |
| Pentachlorophenol |
| Triclosan |
| Emodin |
| Malonoben |
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| cell |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| zebrafish | Danio rerio | High | NCBI |
| human | Homo sapiens | High | NCBI |
| rat | Rattus norvegicus | High | NCBI |
| mouse | Mus musculus | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Embryo | High |
| Juvenile | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
Taxonomic applicability domain
This key event is in general applicable to all eukaryotes, as most organisms are known to use cell proliferation to achieve growth.
Life stage applicability domain
This key event is in general applicable to all life stages. As cell proliferation not only occurs in developing organisms, but also in adults.
Sex applicability domain
This key event is sex-unspecific, as both genders use the same cell proliferation mechanisms.
Key Event Description
Decreased cell proliferation describes the outcome of reduced cell division and cell growth. Cell proliferation is considered the main mechanism of tissue and organismal growth (Conlon 1999). Decreased cell proliferation has been associated with abnormal growth-factor signaling and cellular energy depletion (DeBerardinis 2008).
How it is Measured or Detected
Multiple types of in vitro bioassays can be used to measure this key event:
- ToxCast high-throughput screening bioassays such as “BSK_3C_Proliferation”, “BSK_CASM3C_Proliferation” and “BSK_SAg_Proliferation” can be used to measure cell proliferation status.
- Commercially available methods such as the well-established 5-bromo-2’-deoxyuridine (BrdU) (Raza 1985; Muir 1990) or 5-ethynyl-2’-deoxyuridine (EdU) assay. Both assays measure DNA synthesis in dividing cells to indicate proliferation status.
References
Conlon I, Raff M. 1999. Size control in animal development. Cell 96:235-244. DOI: 10.1016/s0092-8674(00)80563-2.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metabolism 7:11-20. DOI: https://doi.org/10.1016/j.cmet.2007.10.002.
Muir D, Varon S, Manthorpe M. 1990. An enzyme-linked immunosorbent assay for bromodeoxyuridine incorporation using fixed microcultures. Analytical Biochemistry 185:377-382. DOI: https://doi.org/10.1016/0003-2697(90)90310-6.
Raza A, Spiridonidis C, Ucar K, Mayers G, Bankert R, Preisler HD. 1985. Double labeling of S-phase murine cells with bromodeoxyuridine and a second DNA-specific probe. Cancer Research 45:2283-2287.
List of Adverse Outcomes in this AOP
Event: 1521: Decrease, Growth
Short Name: Decrease, Growth
Event Component
| Process | Object | Action |
|---|---|---|
| growth | multicellular organism | decreased |
AOPs Including This Key Event
Stressors
| Name |
|---|
| 2,4-Dinitrophenol |
| Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
| Carbonyl cyanide m-chlorophenyl hydrazone |
| Pentachlorophenol |
| Triclosan |
| Emodin |
| Malonoben |
Biological Context
| Level of Biological Organization |
|---|
| Individual |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human | Homo sapiens | Moderate | NCBI |
| rat | Rattus norvegicus | Moderate | NCBI |
| mouse | Mus musculus | Moderate | NCBI |
| zebrafish | Danio rerio | High | NCBI |
| fathead minnow | Pimephales promelas | High | NCBI |
| Lemna minor | Lemna minor | High | NCBI |
| Daphnia magna | Daphnia magna | Moderate | NCBI |
| Life Stage | Evidence |
|---|---|
| Embryo | High |
| Juvenile | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
Taxonomic applicability domain
This key event is in general applicable to all eukaryotes.
Life stage applicability domain
This key event is applicable to early life stages such as embryo and juvenile.
Sex applicability domain
This key event is sex-unspecific.
Key Event Description
Decreased growth refers to a reduction in size and/or weight of a tissue, organ or individual organism. Growth is normally controlled by growth factors and mainly achieved through cell proliferation (Conlon 1999).
How it is Measured or Detected
Growth can be indicated by measuring weight, length, total volume, and/or total area of a tissue, organ or individual organism.
Regulatory Significance of the AO
Growth is a regulatory relevant chronic toxicity endpoint for almost all organisms. Multiple OECD test guidelines have included growth either as a main endpoint of concern, or as an additional endpoint to be considered in the toxicity assessments. Relevant test guidelines include, but not only limited to:
-Test No. 201: Freshwater Alga and Cyanobacteria, Growth Inhibition Test
-Test No. 208: Terrestrial Plant Test: Seedling Emergence and Seedling Growth Test
-Test No. 211: Daphnia magna Reproduction Test
-Test No. 212: Fish, Short-term Toxicity Test on Embryo and Sac-Fry Stages
-Test No. 215: Fish, Juvenile Growth Test
-Test No. 221: Lemna sp. Growth Inhibition Test
-Test No. 228: Determination of Developmental Toxicity to Dipteran Dung Flies (Scathophaga stercoraria L. (Scathophagidae), Musca autumnalis De Geer (Muscidae))
-Test No. 241: The Larval Amphibian Growth and Development Assay (LAGDA)
-Test No. 407: Repeated Dose 28-day Oral Toxicity Study in Rodents
-Test No. 408: Repeated Dose 90-Day Oral Toxicity Study in Rodents
-Test No. 416: Two-Generation Reproduction Toxicity
-Test No. 422: Combined Repeated Dose Toxicity Study with the Reproduction/Developmental Toxicity Screening Test
-Test No. 443: Extended One-Generation Reproductive Toxicity Study
-Test No. 453: Combined Chronic Toxicity/Carcinogenicity Studies
References
Conlon I, Raff M. 1999. Size control in animal development. Cell 96:235-244. DOI: 10.1016/s0092-8674(00)80563-2.
Appendix 2
List of Key Event Relationships in the AOP
List of Adjacent Key Event Relationships
Relationship: 2009: Increase, ROS leads to Increase, Oxidative Stress
AOPs Referencing Relationship
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
This KER is broadly applicable to aerobic eukaryotic systems in which ROS production and antioxidant buffering can be measured. The current AOP-Wiki relationship page identifies human, mouse and rat with high evidence, but the ROS-growth evidence base supports extension to algae, fish, crustaceans, mollusks and other organisms relevant to environmental toxicology (AOP-Wiki, 2026a). The relationship is expected to be conserved because it is based on redox chemistry and conserved antioxidant-defense systems rather than on a taxon-specific receptor or signaling pathway.
The applicability domain should nevertheless be bounded by biological context and measurement feasibility. This KER is most relevant when the upstream KE is a measurable increase in ROS and the downstream KE is a measurable redox imbalance or antioxidant-response state rather than a distal oxidative damage endpoint alone. In organisms or compartments where ROS cannot be measured directly, evidence may rely on antioxidant-response or oxidative damage biomarkers, but these should be interpreted as indirect support. Applicability is strongest when ROS and oxidative stress endpoints are measured in the same system under the same exposure conditions.
Key Event Relationship Description
This KER describes the causal and predictive relationship by which an increase in reactive oxygen species leads to oxidative stress. ROS include superoxide, hydrogen peroxide, hydroxyl radical and secondary oxygen-derived reactive products. At low or transient levels, ROS can participate in normal cell signaling. However, when ROS production, flux or local concentration exceeds the capacity of enzymatic and non-enzymatic antioxidant defenses, the redox balance of the biological system shifts toward an oxidizing state, producing oxidative stress (Schieber and Chandel, 2014; Sies et al., 2017).
The downstream KE, oxidative stress, is not identical to increased ROS. Rather, it represents a systems-level imbalance between pro-oxidant pressure and antioxidant or repair capacity. The KER therefore depends not only on the magnitude of ROS increase, but also on the duration, localization and chemical identity of the ROS, the capacity of scavenging systems such as glutathione, superoxide dismutase, catalase and glutathione peroxidases, and the ability of the cell or organism to activate adaptive redox responses such as NRF2 signaling (Halliwell and Gutteridge, 2015; Griendling et al., 2016; Sies et al., 2017).
Within the ROS-growth AOP network, Relationship 2009 functions as a shared upstream KER. It connects the early measurable perturbation of increased ROS to the central hub event of oxidative stress, from which downstream AOP branches proceed through oxidative DNA damage, lipid peroxidation, protein oxidation, mitochondrial dysfunction, ATP depletion, altered cell proliferation, cell injury/death and decreased growth. This KER should remain modular and stressor-agnostic; stressor-specific mechanisms of ROS generation should be described in MIE or stressor sections where appropriate.
Evidence Supporting this KER
Biological PlausibilityBiological plausibility of Relationship 2009 is high. ROS are produced endogenously by mitochondrial electron transport, oxidase enzymes, peroxisomal reactions, photosynthetic electron transport and immune-cell oxidant systems, and they may also be generated by redox-cycling chemicals, metals, radiation and other stressors (Bedard and Krause, 2007; Murphy, 2009; Halliwell and Gutteridge, 2015). Oxidative stress is defined as a disturbance in the balance between oxidants and antioxidants in favor of oxidants, leading to disruption of redox signaling and/or molecular damage (Sies et al., 2017). Therefore, a sufficient increase in ROS has a direct mechanistic basis for causing oxidative stress when antioxidant and repair capacity are exceeded.
This relationship is also strongly supported by the known biology of antioxidant defenses. Superoxide dismutases convert superoxide to hydrogen peroxide; catalase, glutathione peroxidases and peroxiredoxins reduce hydrogen peroxide and organic peroxides; and glutathione and thioredoxin systems maintain protein thiol redox balance. Increased ROS can consume these defenses, oxidize redox-sensitive proteins, activate NRF2-dependent antioxidant response pathways, and produce oxidative modification of lipids, proteins and nucleic acids (Schieber and Chandel, 2014; Griendling et al., 2016; Sies et al., 2017).
Empirical EvidenceEmpirical support for this KER is high. Numerous studies across taxa and stressor classes demonstrate concordant increases in ROS or ROS-generating conditions and oxidative stress endpoints. The strongest evidence comes from studies measuring both ROS and antioxidant-response or oxidative-stress biomarkers in the same biological system. Several examples from the ROS-growth concordance table are summarized below.
|
Biological system |
Stressor |
Exposure |
Evidence for KE1115 (ROS increase) |
Evidence for KE1392 (oxidative stress increase) |
Concordance interpretation |
Reference |
|
Chlorella vulgaris |
Paraquat |
24 h; 0-1.0 uM |
DCFH-DA fluorescence increased; LOEC for ROS approximately 0.5 uM paraquat. |
SOD, POD and CAT activities increased at similar concentrations; antioxidant enzymes were approximately 3-5-fold above control at 0.5 uM. |
Dose concordance supports ROS increase leading to oxidative stress in a photosynthetic eukaryote. |
Qian et al. (2009) |
|
Daphnia magna |
Paraquat |
48 h; 0.01-10 uM |
ROS induction threshold reported around 0.1 uM paraquat. |
SOD, CAT and GPx induction observed around 0.5 uM; TBARS increased around 1 uM. |
ROS occurs at lower or similar concentrations than antioxidant and damage markers, supporting dose concordance. |
Barata et al. (2005) |
|
Trachinotus ovatus |
Streptococcus agalactiae infection |
0-120 h; 2 x 10^7 CFU/fish |
ROS increased early, with maximum response around 6 h. |
Antioxidant enzyme activities and antioxidant gene expression changed following the ROS response. |
Temporal concordance supports ROS preceding redox-response activation during pathogen-induced oxidative stress. |
Gao et al. (2022) |
|
Mus musculus |
Copper sulfate |
42 days; 0-40 mg/kg bw |
ROS increased at the lowest tested dose by day 42. |
Antioxidant markers including SOD, GSH-related responses and oxidative stress/inflammatory indicators changed with exposure. |
Concordant ROS and antioxidant-response changes support the relationship in mammals. |
Jian et al. (2020) |
|
Marine bivalves |
Chlorothalonil |
96 h; 0.1-10 ug/L |
Stressor is thiol-reactive and associated with oxidative challenge; direct ROS was not the primary endpoint. |
SOD, CAT and GPx activity changes and MDA/TBARS increases occurred in gill tissues. |
Supports downstream oxidative stress following a stressor known to disturb redox balance; direct ROS evidence is weaker than in rows with ROS measurement. |
Haque et al. (2019) |
|
Mya arenaria |
Cyclic hypoxia/reoxygenation |
3 weeks; repeated low oxygen exposure |
Hypoxia/reoxygenation is a recognized ROS-generating condition in mitochondria. |
Mitochondrial proton leak and oxidative stress-related bioenergetic changes were elevated under cyclic hypoxia. |
Supports environmental modulation of ROS-associated oxidative stress and mitochondrial response. |
Ouillon et al. (2021) |
Uncertainties and Inconsistencies
The main uncertainties relate to measurement specificity and context dependence. ROS are chemically diverse and often short-lived, so different assays may detect different ROS species or generalized oxidant-dependent probe oxidation rather than a single ROS concentration. DCFH-DA and related probes are useful screening tools but can be influenced by peroxidases, metals, light, probe loading and cellular esterase activity (Wardman, 2007; Kalyanaraman et al., 2012). Consequently, apparent ROS increases must be interpreted with assay limitations in mind.
A second uncertainty is that ROS increases are not always adverse. Transient or localized ROS signals may activate adaptive stress responses and restore redox homeostasis without producing sustained oxidative stress. Conversely, oxidative stress may be inferred from antioxidant enzyme induction or oxidative damage biomarkers in studies where ROS were not directly measured. These cases support the KER less strongly than studies with direct, temporally resolved ROS measurements. Differences among taxa, life stages, tissues, exposure durations and antioxidant capacities may alter the threshold at which increased ROS becomes oxidative stress.
Quantitative Understanding of the Linkage
Quantitative understanding of this KER is low to moderate. The qualitative relationship is well established: oxidative stress occurs when ROS production or flux exceeds antioxidant and repair capacity. However, a universal quantitative threshold for ROS leading to oxidative stress cannot be defined because the relationship depends strongly on ROS species, subcellular localization, measurement method, antioxidant capacity, exposure duration, organism, cell type and co-stressors (Kalyanaraman et al., 2012; Griendling et al., 2016; Sies et al., 2017).
Response-response relationshipResponse-response information is available in specific systems. For example, in Chlorella vulgaris exposed to paraquat, ROS and antioxidant enzyme responses were observed at approximately 0.5 uM after 24 h, indicating local dose concordance between the upstream and downstream events (Qian et al., 2009). In Daphnia magna exposed to paraquat, ROS induction was reported at lower concentrations than antioxidant enzyme and TBARS responses, supporting an expected dose sequence in which ROS increases precede oxidative stress endpoints (Barata et al., 2005). These examples provide semi-quantitative support, but they cannot be generalized across all taxa or stressors.
Time-scaleThe time scale of the KER can range from minutes to hours for ROS-sensitive signaling and antioxidant pathway activation, and from hours to days for measurable changes in antioxidant enzyme activities, glutathione status or oxidative damage biomarkers. In pathogen-exposed golden pompano, ROS increased early, followed by antioxidant enzyme and gene expression responses over subsequent hours to days, supporting temporal concordance (Gao et al., 2022).
Known modulating factors|
Modulating factor |
Details |
Effect on the KER |
Supporting evidence |
|
Antioxidant capacity |
Levels and activities of GSH, SOD, CAT, GPx, peroxiredoxins, thioredoxin systems and antioxidant vitamins. |
Higher antioxidant capacity buffers ROS and raises the threshold for oxidative stress; depleted or impaired antioxidant systems lower the threshold. |
Halliwell and Gutteridge (2015); Sies et al. (2017). |
|
NRF2/ARE pathway activation |
Induction of antioxidant and detoxification genes through NRF2-dependent signaling. |
Adaptive NRF2 activation may reduce progression from increased ROS to sustained oxidative stress, but strong NRF2 activation can also serve as evidence that ROS has perturbed redox homeostasis. |
Schieber and Chandel (2014); Sies et al. (2017); AOP-Wiki (2026c). |
|
Subcellular localization of ROS |
Mitochondria, chloroplasts, peroxisomes, membranes, nuclei and phagosomes differ in ROS production and local antioxidant buffering. |
Localized ROS production can cause oxidative stress in a specific compartment even when whole-cell ROS measurements are modest. |
Murphy (2009); Griendling et al. (2016). |
|
Exposure duration and recovery time |
Acute pulses, chronic low-level exposure and repeated stress can produce different redox outcomes. |
Short pulses may be buffered or adaptive; sustained or repeated ROS elevations increase the probability of oxidative stress. |
Sies et al. (2017); Ouillon et al. (2021). |
|
Oxygen availability and hypoxia/reoxygenation |
Oxygen tension affects mitochondrial electron transport and ROS formation. |
Reoxygenation after hypoxia can increase mitochondrial ROS and enhance oxidative stress. |
Ouillon et al. (2021). |
|
Temperature and metabolic rate |
Temperature and metabolic demand alter oxygen flux, mitochondrial activity and antioxidant capacity. |
Higher metabolic activity or thermal stress can increase ROS formation and shift the balance toward oxidative stress. |
Tseng et al. (2011). |
|
Stressor chemistry |
Redox cycling, metal-catalyzed reactions, radiation and mitochondrial inhibition generate ROS by different mechanisms. |
Stressor type influences the ROS species, localization, time course and threshold for oxidative stress. |
Bedard and Krause (2007); Murphy (2009); Qian et al. (2009); Gao et al. (2022). |
Known feedback and feedforward mechanisms influence the linkage. NRF2-dependent antioxidant responses can reduce ROS and restore homeostasis, whereas mitochondrial dysfunction, lipid peroxidation, inflammation and redox-sensitive signaling can amplify ROS generation and sustain oxidative stress. These feedbacks make the KER dynamic and nonlinear, particularly under chronic exposure or repeated stress.
References
AOP-Wiki. 2026a. Relationship 2009: Increase, ROS leads to Increase, Oxidative stress. AOP-Wiki. Available at: https://aopwiki.org/relationships/2009. Accessed 14 May 2026.
AOP-Wiki. 2026b. Event 1115: Increase, Reactive oxygen species. AOP-Wiki. Available at: https://aopwiki.org/events/1115. Accessed 14 May 2026.
AOP-Wiki. 2026c. Event 1392: Increase, Oxidative stress. AOP-Wiki. Available at: https://aopwiki.org/events/1392. Accessed 14 May 2026.
Barata C, Varo I, Navarro JC, Arun S, Porte C. 2005. Antioxidant enzyme activities and lipid peroxidation in the freshwater cladoceran Daphnia magna exposed to redox cycling compounds. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology 140(2):175-186. https://doi.org/10.1016/j.cca.2005.01.013.
Bedard K, Krause KH. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews 87(1):245-313. https://doi.org/10.1152/physrev.00044.2005.
Dickinson BC, Chang CJ. 2011. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nature Chemical Biology 7(8):504-511. https://doi.org/10.1038/nchembio.607.
Esperanza M, Cid A, Herrero C, Rioboo C. 2015. Acute effects of a prooxidant herbicide on the microalga Chlamydomonas reinhardtii: screening cytotoxicity and genotoxicity endpoints. Aquatic Toxicology 165:210-221. https://doi.org/10.1016/j.aquatox.2015.06.004.
Gao J, Liu M, Guo H, Zhu K, Liu B, Liu B, Zhang N, Sun X, Jiang S, Zhang D. 2022. ROS induced by Streptococcus agalactiae activate inflammatory responses via the TNF-alpha/NF-kappaB signaling pathway in golden pompano Trachinotus ovatus (Linnaeus, 1758). Antioxidants 11(9):1809. https://doi.org/10.3390/antiox11091809.
Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A. 2016. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circulation Research 119(5):e39-e75. https://doi.org/10.1161/RES.0000000000000110.
Halliwell B, Gutteridge JMC. 2015. Free Radicals in Biology and Medicine. 5th ed. Oxford: Oxford University Press.
Haque MN, Eom HJ, Nam SE, Shin YK, Rhee JS. 2019. Chlorothalonil induces oxidative stress and reduces enzymatic activities of Na+/K+-ATPase and acetylcholinesterase in gill tissues of marine bivalves. PLoS ONE 14(4):e0214236. https://doi.org/10.1371/journal.pone.0214236.
Jian Z, Guo H, Liu H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L. 2020. Oxidative stress, apoptosis and inflammatory responses involved in copper-induced pulmonary toxicity in mice. Aging 12(17):16867-16886. https://doi.org/10.18632/aging.103585.
Kalyanaraman B, Darley-Usmar V, Davies KJA, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ II, Ischiropoulos H. 2012. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radical Biology and Medicine 52(1):1-6. https://doi.org/10.1016/j.freeradbiomed.2011.09.030.
Murphy MP. 2009. How mitochondria produce reactive oxygen species. Biochemical Journal 417(1):1-13. https://doi.org/10.1042/BJ20081386.
Ouillon N, Sokolov EP, Otto S, Rehder G, Sokolova IM. 2021. Effects of variable oxygen regimes on mitochondrial bioenergetics and reactive oxygen species production in a marine bivalve, Mya arenaria. Journal of Experimental Biology 224(4):jeb237156. https://doi.org/10.1242/jeb.237156.
Pan YX, Luo Z, Zhuo MQ, Wei CC, Chen GH, Song YF. 2018. Oxidative stress and mitochondrial dysfunction mediated Cd-induced hepatic lipid accumulation in zebrafish Danio rerio. Aquatic Toxicology 199:12-20. https://doi.org/10.1016/j.aquatox.2018.03.017.
Qian H, Chen W, Sun L, Jin Y, Liu W, Fu Z. 2009. Inhibitory effects of paraquat on photosynthesis and the response to oxidative stress in Chlorella vulgaris. Ecotoxicology 18(5):537-543. https://doi.org/10.1007/s10646-009-0311-8.
Schieber M, Chandel NS. 2014. ROS function in redox signaling and oxidative stress. Current Biology 24(10):R453-R462. https://doi.org/10.1016/j.cub.2014.03.034.
Sies H, Berndt C, Jones DP. 2017. Oxidative stress. Annual Review of Biochemistry 86:715-748. https://doi.org/10.1146/annurev-biochem-061516-045037.
Tseng YC, Chen RD, Lucassen M, Schmidt MM, Dringen R, Abele D, Hwang PP. 2011. Exploring uncoupling proteins and antioxidant mechanisms under acute cold exposure in brains of fish. PLoS ONE 6(3):e18180. https://doi.org/10.1371/journal.pone.0018180.
Wardman P. 2007. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radical Biology and Medicine 43(7):995-1022. https://doi.org/10.1016/j.freeradbiomed.2007.06.026.
Relationship: 2810: Increase, Oxidative Stress leads to Increase, Oxidative DNA damage
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Deposition of energy leading to occurrence of cataracts | adjacent | Moderate | Low |
| Excessive reactive oxygen species leading to growth inhibition via oxidative DNA damage | adjacent | ||
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell cycle disruption | adjacent | High | Moderate |
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell death | adjacent | High | Moderate |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages | Moderate |
| Sex | Evidence |
|---|---|
| Unspecific | Moderate |
This KER is plausible in all life stages, sexes, and organisms with DNA. The majority of the evidence is from in vivo studies conducted in male and female adult mice and rats. No in vitro evidence was found to support the relationship.
Key Event Relationship Description
Oxidative stress refers to a state in which the amount of reactive oxygen (ROS) and nitrogen (RNS) species overwhelms the cell’s antioxidant defense system. This loss in redox homeostasis can lead to oxidative damage to proteins, lipids, and nucleic acids (Schoenfeld et al., 2012; Tangvarasittichai & Tangvarasittichai, 2018; Turner et al., 2002). ROS are molecules with oxygen as the functional center and at least one unpaired electron in the outer orbits. Although less common than ROS, RNS can also induce oxidative stress (Cadet et al., 2012; Tangvarasittichai & Tangvarasittichai, 2018).
Organisms contain a defense system of antioxidants to help manage ROS levels. Antioxidant measures consist of antioxidant enzymes, vitamins and minerals that catalyze the conversion of ROS to non-toxic molecules such as water and O2. When an antioxidant system is overwhelmed by the amount of ROS, the cell can enter a state of oxidative stress (Balasubramanian, 2000; Ganea & Harding, 2006; Karimi et al., 2017).
Unmanaged oxidative stress can damage vital macromolecules such as DNA leading to oxidative DNA damage. This can be divided into two categories, damage caused by one ROS, and damage caused by at least two ROS associating with the DNA in the space of one to two helix turns. The first scenario initiates DNA-protein cross-links, inter and intrastrand links, and tandem base lesions, while the second scenario produces more complicated lesions, known as oxidatively generated clustered lesions (ODCLs). These can include single and double strand breaks, abasic sites, and oxidized bases (Cadet et al., 2012) which can cause chromosomal aberrations, cytotoxicity, and oncogenic transformations (Stohs, 1995) as well as structural changes to the DNA, such as blocking polymerases (Zhang et al., 2010).
8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodG) lesions are the most common and best-studied, as such they are often used as a marker of oxidative DNA damage (Tangvarasitichai & Tangvarasittichai, 2018).
Cells possess DNA repair mechanisms that help repair the damage, but these processes are not perfect (Eaton, 1995; Ainsbury et al., 2016; Markkanen, 2017). Furthermore, certain types of lesions, such as DNA double strand breaks, are more complex to repair (Schoenfeld et al, 2012), leading to increased oxidative DNA damage.
Evidence Supporting this KER
Overall Weight of Evidence: Moderate
Biological PlausibilityWhen a cell is exposed to oxidative stress, DNA lesions can be induced. There are various repair systems that will attempt to repair the damage sometimes successfully, and other times inadequately or inefficiently, in this case oxidative DNA damage will persist. Furthermore, if there are too many lesions, the DNA repair system may be overwhelmed. A low level of damage is always found in healthy cells, but this amount increases under oxidative stress (Lee et al., 2004). It has been estimated that human cells have 70 000 lesions per day, mostly due to ROS produced during normal metabolism and base hydrolysis (Amente et al., 2019). These lesions can be DNA breaks, but there are also other types such as oxidized bases. Furthermore, while ROS induces DNA breaks, it can also be caused by other processes, or be an intermediate in DNA repair. As a result, oxidized nucleotides are generally a more accurate indicator of oxidative stress (Collins, 2014).
Oxidative stress affects different nitrogenous bases differently. For example, guanine (G) has a lower redox potential, causing it to be more vulnerable to oxidation compared to other nitrogenous bases. This leads to increased amounts of oxidized G products, relative to other forms of damage, Furthermore, ribonucleotides can also be oxidized, to the point where dGTP is more vulnerable to oxidation than G (Markkanen, 2017). Certain compounds such as hydroxyl radical generation systems and adriamycin-iron complexes will bind to and form ROS in association with DNA, therefore inducing site-specific DNA damage (Stohs, 1995).
Additionally, cells that are actively dividing are more sensitive to oxidative DNA damage (Sacca et al., 2009). A few studies have also found that single stranded DNA (ssDNA) is more likely to be oxidized than double stranded DNA (dsDNA). This indicates that persistent ssDNA sites, such as Z-DNA, stable R-loops, cruciforms, quadruplexes, or intramolecular triplexes might have higher incidences of oxidative damage (Amente et al., 2019).
Cells use three main methods to repair and prevent oxidative DNA damage. Firstly, enzymes such as Mut homologue 1, 2, 3, and Nudix-type 5 (MTH1, MTH2, MTH3, and NUDT5) are used to remove oxidized nucleotides before they can be incorporated into DNA. Another method is switching between replicative polymerases and DNA polymerase γ (Polγ) during replication when an 8-oxo-G lesion is encountered. This allows the replicative machinery to bypass the lesion. The third method is the base excision repair (BER) pathway, which is the major DNA repair pathway for base damage and has two general sub paths. The first is the short patch, where only the damaged nucleotides are replaced. The other is the long patch, which replaces a group of 2 to 12 nucleotides (Markkanen, 2017). For mitochondrial DNA (mtDNA), which is more sensitive to oxidative damage than nuclear DNA (Yakes & Van Houten, 1997), BER involves three main enzymes. 8-oxoguanine DNA glycosylase 1 (OGG1) removes 8-OHdG lesions, which are caused by the incorporation of 8-oxodGTP. AP endonuclease 1 (APE1) is an AP endonuclease that increases OGG1 turnover and adds a nick to the DNA, preparing it for further repair processes. Finally, DNA polymerase γ (Polγ) adds new nucleotides where the older ones were removed (Zhang et al., 2010). Another kind of BER pathway is SSBR (single strand break repair). When two SSBs are in juxtaposition, they can form DSBs, which are detrimental (Caldecott, 2024; Pfeiffer et al., 2000).
Different lesions are also repaired differently and can cause varying amounts of damage. For example, DNA single strand breaks are usually repaired quickly (Collins, 2014), while double strand breaks are more complicated and are therefore, less likely to be repaired correctly (Schoenfeld et al, 2012). More details on these processes are reviewed in Markkanen (2017). Overall the mechanism to oxidative stress leading to oxidative DNA damage is well accepted and understood.
Empirical EvidenceThere is limited evidence supporting time- or dose-concordance.
Dose Concordance
Zhang et al. (2010) exposed male rats in vivo to 10%, 21% (atmospheric level) and 60% O2 (to induce oxidative stress). This resulted in a 1.5x increase in 8-OHdG levels. It was assumed that 60% oxygen induced oxidative stress, however the study only measured the downstream KE.
Time Concordance
Although DNA damage induced by oxidative stress can be repaired rapidly, the accumulation of oxidative stress typically causes oxidative DNA damage after several months. Two studies show an increase in damage 1.5 and two months respectively after the induction of oxidative stress (Pendergrass et al., 2010 – 2.5x increase in 8-OH-dG positive DNA fragments, in vivo irradiation with 11 Gy X-rays at 2 Gy//min) (Zhang et al., 2010 – 1.6x increase in 8-OHdG, exposure to 60% O2).
Pendergrass et al. (2010) reported that the amount of oxidative DNA damage increased as the amount of time after irradiation increased. It was observed that DNA damage (represented by the number of nuclear fragments in the lens cortex after exposure to 11 Gy X-rays) increased from 100 to 750 fragments from the time of radiation to over 22 months after. It was also shown that the amount of 8-OH G positive DNA fragments increased from about 5 to 55 from the time of radiation (11 Gy X-rays) to 11 months post-exposure (Pendergrass et al., 2010).
Essentiality
No evidence.
Uncertainties and InconsistenciesNo evidence.
Quantitative Understanding of the Linkage
Available data suggests that increases in oxidative stress leads to increases in oxidative DNA damage. The following tables provide representative examples of the relationship, unless otherwise indicated, all data is significantly significant.
Dose Concordance
|
Reference |
Experiment Description |
Result |
|
Zhang et al., 2010 |
In vivo. 72 male Wistar rats were exposed to 21%, and 60% O2 to induce oxidative stress. Oxidative DNA damage was measured by determining 8-hydroxy-2’-deoxy-guanosine (8-OHdG) via competitive ELISA assays. |
In rats exposed in vivo, a 39% increase in atmospheric O2 concentration (indicative of oxidative stress) resulted in a 1.27x increase in 8-OHdG. |
Time Concordance
|
Reference |
Experiment Description |
Result |
|
Pendergrass et al., 2010
|
In vivo. Female, 3-month-old, C57BL/6 mice had their heads exposed to 11 Gy X-rays at 2 Gy/min to induce oxidative stress. Oxidative DNA damage was measured using antibody staining of fixed eyes and immunofluorescence. |
In mice exposed in vivo to 11 Gy X-rays, oxidative stress increased 4.3x relative to control 6 months post-irradiation. The amount of 8-OH G positive DNA fragments increased to 2.7x control 6.5 months after the increase in oxidative stress. |
| Modulating Factor (MF) | MF Specification | Effect(s) on the KER | Reference(s) |
|---|---|---|---|
| Age | Increased age | Increased levels of oxidative DNA damage, partly due to decreased antioxidant levels, meaning that the removal of ROS occurs more slowly, increasing the level of oxidative damage. Moreover, in humans, after about forty to fifty years, a barrier forms in the lens of the eye that decreases intracellular antioxidant transportation. Normally, antioxidants circulate via a current in the cytoplasm of lens fiber cells. However, as the age of the organism increases, the cytoplasm of these cells becomes stiffer. Small molecules such as H2O2 and the superoxide anion can diffuse through, but larger molecules, such as glutathione, cannot enter the barrier. As a result, the core of the lens has a decreased antioxidant concentration, making it more vulnerable to oxidative damage. Furthermore, the amount of protein and mRNA corresponding to important mitochondrial BER enzymes decreases with age, causing a decrease in DNA repair ability and therefore an increase in DNA damage in the mitochondria. | Stohs, 1995; Lee et al., 2004; Martinez et al., 2010; Pendergrass et al., 2010; Zhang et al., 2010; Ainsbury et al., 2016; Tangvarasittichai & Tangvarasittichai, 2018 |
| H2 | Increased concentration | Decreased level of oxidative DNA damage. | Schoenfeld et al., 2012 |
| Antioxidants | Increased concentration | Reviews have found that about 50% of studies examined showed a decrease in base oxidation, but the other half show no change. | Turner et al., 2002; Møller & Loft, 2006; Hoelzl et al., 2009 |
| Lipoic acid | Increased concentration | Decreased level of oxidative DNA damage. | Turner et al., 2002 |
| Acetyl carnitine | Increased concentration | Decreased level of oxidative DNA damage. | Turner et al., 2002 |
| Ubiquinone Q-9 | Increased concentration | Decreased level of oxidative DNA damage. | Turner et al., 2002 |
| Hydroquinone | Increased concentration | Decreased level of oxidative DNA damage. | Turner et al., 2002 |
| Folate | Increased concentration | Decreased level of oxidative DNA damage. | Turner et al., 2002 |
| Aged garlic extracts | Increased concentration | Decreased level of oxidative DNA damage. | Turner et al., 2002 |
Not identified
References
Ainsbury, E. A. et al. (2016), “Ionizing radiation induced cataracts: Recent biological and mechanistic developments and perspectives for future research”, Reviews in mutation research, Vol. 770, Elsevier. https://doi.org/10.1016/j.mrrev.2016.07.010
Amente, S. et al. (2019), “Genome-wide mapping of 8-oxo-7,8-dihydro-2’-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells”, Nucleic Acids Research 2019, Vol. 47/1, Oxford University Press, England, https://doi.org/10.1093/nar/gky1152
Cadet, J. et al. (2012), “Oxidatively generated complex DNA damage: tandem and clustered lesions”, Cancer letters, Vol. 327/1, Elsevier Ireland Ltd, Ireland, https://doi.org/10.1016/j.canlet.2012.04.005
Cadet, J., and& Davies, K.J. A (2017). “Oxidative DNA damage & repair: An introduction. Free radical biology & medicine” Vol.107, 2-12 https://doi.org/10.1016/j.freeradbiomed.2017.03.030
Caldecott K. W. (2024). “Causes and consequences of DNA single-strand breaks”. Trends in biochemical sciences, Vol 49/1, 68–78. https://doi.org/10.1016/j.tibs.2023.11.001.
Collins, A. R. (2014), “Measuring oxidative damage to DNA and its repair with the comet assay”, Biochimica et biophysica acta. General subjects, Vol. 1840/2, Elsevier B.V., https://doi.org/10.1016/j.bbagen.2013.04.022
Eaton, J. W. (1995), “UV-mediated cataractogenesis: a radical perspective”, Documenta ophthalmologica, Vol. 88/3-4, Springer, Dordrecht, https://doi.org/10.1007/BF01203677
Hoelzl, C. et al. (2009), “Use of single cell gel electrophoresis assays for the detection of DNA-protective effects of dietary factors in humans: Recent results and trends”, Mutation Research, Vol. 681/1, Elsevier, https://doi.org/10.1016/j.mrrev.2008.07.004
Kozbenko, T. et al. (2022), “Deploying elements of scoping review methods for adverse outcome pathway development: a space travel case example”, International Journal of Radiation Biology, 1–12. https://doi.org/10.1080/09553002.2022.2110306
Lee, J., N, Koo and D. B. Min (2004), “Reactive oxygen species, aging, and antioxidative nutraceuticals”, Comprehensive reviews in food science and food safety, Vol. 3/1, Blackwell Publishing Ltd, Oxford, https://doi.org/10.1111/j.1541-4337.2004.tb00058.x
Markkanen, E. (2017), “Not breathing is not an option: How to deal with oxidative DNA damage”, DNA repair, Vol. 59, Elsevier B.V., Netherlands, https://soi.org/10.1016/j.dnarep.2017.09.007
Martinez, G. and R. U. de Longh (2010), “The lens epithelium in ocular health and disease”, The international journal of biochemistry & cell biology, Vol. 42/12, Elsevier B. V, Netherlands, https://doi.org/10.1016/j.biocel.2010.09.012
Møller, P. and Loft, S. (2006), “Dietary antioxidants and beneficial effect on oxidatively damaged DNA”, Free Radical Biology and Medicine, Elsevier, https://doi.org/10.1016/j.freeradbiomed.2006.04.001
Pendergrass, W. et al. (2010), “X-ray induced cataract is preceded by LEC loss, and coincident with accumulation of cortical DNA, and ROS; similarities with age-related cataracts”, Molecular vision, Vol. 16, Molecular Vision, United States, pp. 1496-1513
Pfeiffer, P. et al. (2000),. “Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations”,. Mutagenesis, Vol.15(4). https://doi.org/10.1093/mutage/15.4.289
Sacca, S. C. et al. (2009), “Gene-environment interactions in ocular diseases”, Mutation research – fundamental and molecular mechanisms of mutagenesis, Vol. 667/1-2, Elsevier, Amsterdam, https://doi.org/10.1016/j.mrfmmm.2008.11.002
Schoenfeld, M. P. et al. (2012), “A hypothesis on biological protection from space radiation through the use of new therapeutic gases as medical counter measures”, Medical gas research, Vol. 2/1, BioMed Central Ltd, India, https://doi.org/10.1186/2045-9912-2-8
Stohs, S. J. (1995), “The role of free radicals in toxicity and disease”, Journal of Basic and Clinical Physiology and Pharmacology, Vol. 6/3-4, Freund Publishing House Ltd, https://doi.org/10.1515/JBCPP.1995.6.3-4.205
Tangvarasittichai O. and S. Tangvarasittichai (2018), “Oxidative stress, ocular disease and diabetes retinopathy”, Current Pharmaceutical Design, Vol. 24/40, https://doi.org/10.2174/1381612825666190115121531
Turner, N. D. et al. (2002), “Opportunities for nutritional amelioration of radiation-induced cellular damage”, Nutrition, Vol. 18/10, Elsevier Inc, New York, http://doi.org/10.1016/S0899-9007(02)00945-0
Yakes, F. M. and B. Van Houten (1997), “Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress”, Cell Biology, Vol. 94, The National Academy of Sciences of the USA, United States, pp. 514-519
Zhang, Y. et al. (2010), “Oxygen-induced changes in mitochondrial DNA and DNA repair enzymes in aging rat lens”, Mechanisms of ageing and development, Vol. 131/11, Elsevier Ireland Ltd, Clare, https://doi.org/10.1016/j.mad.2010.09.003
Relationship: 1909: Increase, Oxidative DNA damage leads to Inadequate DNA repair
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Oxidative DNA damage leading to chromosomal aberrations and mutations | adjacent | High | Low |
| Deposition of energy leading to occurrence of cataracts | adjacent | Moderate | Low |
| Excessive reactive oxygen species leading to growth inhibition via oxidative DNA damage | adjacent | ||
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell cycle disruption | adjacent | High | Low |
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell death | adjacent | High | Low |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages | Moderate |
| Sex | Evidence |
|---|---|
| Unspecific | Moderate |
This KER is plausible in all life stages, sexes, and organisms with DNA. The majority of the evidence is from in vivo mice studies of all ages with no specification on sex. No in vitro evidence was found to support the relationship.
Key Event Relationship Description
Oxidative DNA lesions are present in the cell at steady state due to low levels of reactive oxygen species (ROS) and other free radicals generated by endogenous processes involving redox reactions. The most prominent examples of oxidative DNA lesions include 7, 8-dihydro-8oxo-deoxyGuanine (8-oxo-dG), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FaPydG), and thymidine glycol (Tg). Under homeostatic conditions, cells are able to regulate the level of free radicals and readily repair oxidized DNA bases using basal repair mechanisms to prevent irreversible damage (Swenberg et al., 2011). Oxidative DNA lesions are mainly repaired by base excision repair (BER) initiated by DNA glycosylases such as oxoguanine glycosylase 1 (OGG1), endonuclease III homologue 1 (NTH1), and Nei-like DNA glycosylases (NEIL 1/2), which detect and remove damaged bases. Abasic sites are then cleaved by endonucleases or lyases, resulting in transient single-strand breaks (SSB) that enter either short-patch or long-patch repair. Nucleotide excision repair (NER) and single-strand base repair is also involved in repairing oxidized bases to a lesser extent (Shafirovich et al., 2016; Hedge et al., 2012). Increase in free radicals or exposure to oxidizing agents can increase the level of oxidative DNA lesions and overwhelm the repair pathways, compromising the quality of repair. If the repair mechanisms are compromised, oxidative lesions may accumulate (insufficient repair) and cause incorrect base pairing during replication or incomplete repair (indicated by accumulation of repair intermediates) (Markkanen, 2017).
Evidence Supporting this KER
Overall Weight of Evidence: Moderate
Inadequate repair of oxidative lesions is indicated by an increase in oxidative lesions above background, activation of repair enzymes, increase in repair intermediates (abasic sites and SSBs), and incorrect base insertion opposite lesion during replication (lesion bypass by translesion DNA synthesis).
Biological PlausibilityThe mechanism of repair of oxidative DNA lesions in humans is well-established and numerous literature reviews are available on this topic (Berquist and Wilson III, 2012; Cadet and Wagner, 2013). As described above, oxidative DNA lesions are mostly repaired via BER and, to a lesser extent, NER. Previous studies have reported thresholded dose-response curves in oxidative DNA damage and attributed these observations to exceeded repair capacity at the inflection point on the curve (Gagne et al., 2012; Seager et al., 2012). In vivo, increase and accumulation of oxidative DNA lesions despite the activation of BER have been observed following chemical exposures, demonstrating insufficient repair of oxidative DNA lesions past a certain level (Ma et al., 2008).
OGG1 and NTH1, the glycosylases that initiate the BER of 8-oxo-dG and thymine glycol (Tg) lesions, respectively, are bifunctional, containing both glycosylase and lyase activities. The glycosylase removes the oxidized guanine by cleaving the glycosidic bond, giving rise to an apurinic site. The lyase then cleaves the phosphodiester bond 5’ to the AP site; a transient SSB is created for further processing in BER (Delaney et al., 2012). Abasic sites created by OGG1 and other glycosylases are also processed by apuric/apyrimidinic endonucleases (APE1) to create the 5’ nick (Allgayer et al., 2016). The repair process can be inhibited when non-DSB oxidative DNA damage results in altered nuclease or glycosylase activity, making the area resistant to repair following radiation exposure (Georgakilas et al., 2013).
Previous studies have demonstrated that an imbalance in any one of the multiple steps of BER can lead to an accumulation of repair intermediates and failed repair. Given that OGG1 is relatively slower in releasing its catalytic product than other glycosylases, it is highly likely that a disproportionate increase in oxidative DNA lesions compared to the level of available OGG1 would lead to an imbalance between lesions and the initiating step of BER (Brenerman et al., 2014). Accumulation of oxidative lesions would be observed as a result. Moreover, studies have reported accumulation of SSB due to OGG1 and NTH1 overexpression, demonstrating that the imbalanced lyase activity generates excessive SSB intermediates (Yang et al., 2004; Yoshikawa et al., 2015; Wang et al., 2018).
Increases in oxidative lesions may produce more lesions and repair intermediates in close proximity to each other. Previous studies in mammalian cell extracts have reported reduction in repair efficiency when oxidative lesions are in tandem or opposite each other. For example, OGG1 showed reduced binding to 8-oxo-dG near an AP site incision. Furthermore, the OGG1-8-oxo-dG complex has been observed to hinder the repair of neighbouring AP site incision, delaying the completion of BER; this interaction between BER enzymes has been suggested to cause an accumulation of oxidative lesions and repair intermediates (Pearson et al., 2004; Budworth et al., 2005; Bellon et al., 2009; Yoshikawa et al., 2015; Sharma et al., 2016; Georgakilas et al., 2013).
If oxidative lesions persist in the genome due to insufficient repair, incorrect base insertion opposite unrepaired oxidative DNA lesions may occur during replication. This is a well-established event. For example, 8-oxo-dG and FaPydG, the two most prominent oxidative DNA lesions, are able to form base pairs with dATP, giving rise to G:C→T:A transversions after subsequent DNA synthesis (Freudenthal et al., 2013; Gehrke et al., 2013; Markkanen, 2017). Replicative DNA polymerases such as DNA polymerase α, δ, and ε (pol α, δ, ε) have a poor ability to extend the DNA strand past 8-oxo-dG:dCTP base pairs and may cause replication to stall or incorrectly insert dATP opposite 8-oxo-dG (Hashimoto et al., 2004; Markkanen et al., 2012). In stalled replication forks, repair polymerases may be recruited to perform translesion DNA synthesis (TLS). Human Y-family DNA polymerases (Rev 1, pol κ, ι, and η) are DNA repair polymerases mainly involved in TLS in stalled replication forks. However, TLS is not free of error and its accuracy differs for each repair polymerase. For example, it is known that pol κ and η perform TLS across 8-oxo-dG and preferentially insert dATP opposite the lesion, generating G:C→T:A transversions. The error-prone nature of bypassing unrepaired oxidative lesions has been described in many previous studies and reviews (Greenberg, 2012; Maddukuri et al., 2014; Taggart et al., 2014; Shah et al., 2018). There is also risk associated with repairing the lesions, that the process could lead to increased genomic instability and mutation potential. A balance needs to be achieved between the risk posed by repair and that by residual oxidative damage (Poetsch, 2020).
Repair by OGG1 requires 8-oxo-dG:dC base pairing, thus, it is unable to repair 8-oxo-dG:dA mispairing in newly synthesized strands. The repair of 8-oxo-dG:dA base pairs post-replication is performed by MUT Y homologue, MYH, an adenine DNA glycosylase. However, the removal of dA instead of the damaged guanine may lead to futile cycles of BER because: 1) another dA is often inserted opposite the lesion, or 2) BER ligases have a poor ability of ligating the 3’end of dC opposite 8-oxo-dG (Hashimoto et al., 2004; Caglayan and Wilson, 2015). Accumulated 8-oxo-dG may be more resistant to repair post-replication due to this futile BER.
Empirical EvidenceExample in vitro studies demonstrating dose and temporal concordance, or essentiality
- Human normal hepatocytes (HL-7702) were exposed to N,N-dimethylformamide for 24 hours at increasing concentrations (C. Wang et al., 2016)
- Concentration-dependent increase in ROS was observed; the increase was statistically significant compared to control at all concentrations (6.4, 16, 40, 100 mM)
- No significant increase in 8-oxodG was observed until the highest two concentrations (40 and 100 mM) indicating insufficient repair at these concentrations
- Significant up-regulation of excision repair genes (XRCC2 and XRCC3) occurred at 6.4 and 16 mM, below the concentrations that significantly induced 8-oxodG, supporting sufficient DNA repair at these low concentrations.
- These results demonstrate that repair is sufficient at low concentrations (rapidly removing 8-oxodG) and not until higher concentrations is repair overwhelmed (i.e., insufficient), where 8-oxo-dG significantly increases.
- AS52 Chinese hamster ovary cells (wild type and OGG1-overexpressing (OGG1+)) were exposed to varying doses of ultraviolet A (UVA) radiation (Dahle et al., 2008)
- Formamidopyrimidine glycosylase (Fpg)-sensitive sites were quantified using alkaline elution after increasing repair times (0, 1, 2, 3, 4 h) following 100 kJ/m2 UVA irradiation
- OGG1-overexpressing AS52 cells (OGG1+): Fpg-sensitive sites reduced to 71% within half an hour and down to background levels at 4h
- Wild type AS52 cells: at 4h, 70% of the Fpg-sensitive sites remained, indicating accumulation of oxidative lesions
- The above results demonstrated that excess OGG1 was able to prevent the accumulation of oxidative lesions, while the amount of OGG1 in wild type was insufficient to handle the amount of lesions induced by the same magnitude of UVA irradiation.
- Mutations in the Gpt gene was quantified in both wild type and OGG1+ cells by sequencing after 13-15 days following 400 kJ/m2 UVA irradiation
- G:C→T:A mutations in UVA-irradiated OGG1+ cells were completely eliminated (thus, repair was sufficient when repair overexpressed).
- G:C→T:A mutation frequency in wild type cells increased from 1.8 mutants/million cells to 3.8 mutants/million cells following irradiation – indicating incorrect repair or lack of repair of accumulated 8-oxo-dG.
- The above result also demonstrates the essentiality of 8-oxo-dG formation in the oxidative DNA damage-induced G to T transversion mutations.
- HL-60 human leukemia cells were irradiated with X-rays at a rate of 0.5 Gy/min for increasing durations (i.e., increasing doses). 8-OHdG levels were quantified by HPLC as number of 8-OHdG per 106 deoxyguanosine (Li et al., 2013)
- No increase in 8-OHdG was observed up to 2 Gy (sufficient repair at low doses), above which the level of lesions increased linearly up to 20 Gy (insufficient repair)
- This thresholded dose-response curve, indicative of overwhelmed repair processes, was also observed in mouse liver in the same study described below.
In vivo studies demonstrating dose or time concordance
- Two groups of 5-week-old C57BL/6J mice were exposed to increasing doses of X-rays at a rate of 0.5 Gy/min (200 kV, 12 mA). The livers were collected from one group immediately after exposure and urine samples were collected over 24 hours following irradiation in the second group of mice (Li et al., 2013).
- 8-OHdG in the mouse liver DNA were quantified by HPLC and expressed as 8-OHdG per 106 deoxyguanosine
- Between 0 and 0.5 Gy, no increase in lesions was observed
- Between 0.5 and 30 Gy, a linear dose-response in 8-OHdG was observed
- The thresholded dose-response curve was concordant in the urine samples; no increase in urinary 8-OHdG (8-OHdG/creatinine (ng/mg)) was observed between 0 and 0.1 Gy but between 0.1 and 5 Gy, the number of lesions increased linearly with dose
- Male Sprague-Dawley rats were fed 0.5 mmol aniline/kg/day for 30 days. Genomic DNA, nuclear extracts, and mitochondrial extracts were collected from spleen tissues (Ma et al., 2008).
- 8-OHdG was quantified using enzyme-linked immunosorbent assay (ELISA) on digested genomic DNA. There was a significant 2.8-fold increase in lesions in aniline-fed rats than in control rats.
- Both the nuclear extracts and mitochondrial extracts were tested for OGG1 activity, where 1.32-fold and 1.15-fold increase in enzyme activity (both significant; p<0.05) were observed in the respective extracts of aniline-treated rats.
- The OGG1 enzyme content in the extracts was detected using Western blotting; the increase in OGG1 content in aniline-treated rats was consistent with the OGG1 activity assay.
- Despite the increase in OGG1 enzyme content and activity, the quantity of 8-OHdG increased.
- Together, these results demonstrate that repair is sufficient at low concentrations because 8-oxodG adducts are rapidly removed. At higher concentrations, 8-oxo-dG begins to significantly increase indicating repair is overwhelmed (i.e., insufficient).
-
Two groups of C57BL/6J mice received lens-specific irradiation in vivo with 3 mJ/cm2 UVB a week apart, with one group being sacrificed 7 days after exposure and the other sacrificed immediately. Immunofluorescence was used to observe cyclobutane pyrimidine dimers (CPD) (Mesa & Bassnett, 2013).
-
Exposed lenses showed a 25% decrease in cyclobutane pyrimidine dimer levels seven days post-exposure.
-
Although the dual functionality of OGG1 as a glycosylase and lyase has been widely accepted and demonstrated experimentally, there are studies showing that the cleavage of phosphodiester bond 5’ to the lesion is mainly performed by apurinic endonuclease 1 (APE1) (Allgayer et al., 2016; R. Wang et al., 2018). In some cases, APE1 may be the main factor driving the accumulation of BER intermediates. Some studies suggest that OGG1 is involved in the repair of non-transcribed strands and is not required for transcription-coupled repair of 8-oxo-dG; Le Page et al. reported efficient repair of 8-oxo-dG in the transcribed sequence in Ogg1 knockout mouse cells (Le Page et al., 2000). Moreover, the repair of 8-oxo-dG is also affected by the neighbouring sequence; the position of the lesions may have a negative effect on repair efficiency (Pastoriza-Gallego et al., 2007). We note that the study by Allgayer et al. was investigating the fate and effect of 8-oxo-dG during transcription; repair mechanism may vary by situation and availability of repair enzymes at the time.
Quantitative Understanding of the Linkage
The precise relationship between levels of oxidative DNA lesions and when repair can be considered inadequate have not been fully defined; this relationship will very likely differ between cell types and tissues and, thus, difficult to define. There are computational models of repair kinetics of 8-oxo-dG.
Sokhansanj and Wilson III [2004] applied a quantitative model of BER and the literature value for the rate of formation of endogenous 8-oxo-dG to investigate the rate of clearance of BER repair intermediates (Sokhansanj and Wilson III, 2004).
- The BER model used Michaelis-Menten enzyme kinetics and included the activities of OGG1, AP lyases, polymerases, and ligases.
- The model assumed the formation rate of endogenous oxidative lesions to be 500 8-oxo-dG/day
- Based on the above, it was estimated that following a sudden spike in 8-oxo-dG up to 20,000 8-oxo-dG/cell, the total level of repair intermediates would return to baseline within 4000 seconds (less than 1 hour)
- This model also assumed that OGG1 was available in excess
- When APE1 (AP site endonuclease) is present, glycosylase reaction kinetics of OGG1(a bifunctional glycosylase/lyase) was observed to increase
- Suggested to be due to the coordinated action of the two enzymes
- A 10-fold reduction in OGG1 kinetics led to 10-fold increase in 8-oxo-dG, while no other repair intermediates increased.
| Modulating Factor (MF) | MF Specification | Effect(s) on the KER | Reference(s) |
|---|---|---|---|
N/A
References
Allgayer, J., Kitsera, N., Bartelt, S., Epe, B., Khobta, A. (2016), Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine, Nucleic Acids Res, 44:7267-7280.
Bellon, S., Shikazono, N., Cunniffe, S., Lomax, M., O’Neill, P. (2009), Processing of thymine glycol in a clustered DNA damage site: mutagenic or cytotoxic, Nucleic Acids Res, 37:4430-4440.
Berquist, B., Wilson III, D. (2012), Pathways for Repairing and Tolerating the Spectrum of Oxidative DNA Lesions, Cancer Lett, 327:61-72.
Brenerman, B., Illuzzi, J., Wilson III, D. (2014), Base excision repair capacity in informing healthspan, Carcinogenesis, 35:2643-2652.
Budworth, H., Matthewman, G., O’Neill, P., Dianov, G. (2005), Repair of Tandem Base Lesions in DNA by Human Cell Extracts Generates Persisting Single-strand Breaks, J Mol Biol, 351:1020-1029.
Cadet, J., Wagner, J.R. (2013), DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation, Cold Spring Harb Perspect Biol, 5:a012559.
Caglayan, M., Wilson, S. (2015), Oxidant and environmental toxicant-induced effects compromise DNA ligation during base excision DNA repair, DNA Repair, 35:85-89.
Dahle, J., Brunborg, G., Svendsrud, D., Stokke, T., Kvam, E. (2008), Overexpression of human OGG1 in mammalian cells decreases ultraviolet A induced mutagenesis, Cancer Lett, 267:18-25.
Delaney, S., Jarem, D., Volle, C., Yennie, C. (2012), Chemical and Biological Consequences of Oxidatively Damaged Guanine in DNA, Free Radic Res, 46:420-441.
Freudenthal, B., Beard, W., Wilson, S. (2013), DNA polymerase minor groove interactions modulate mutagenic bypass of a templating 8-oxoguanine lesion., Nucleic Acids Res, 41:1848-1858.
Gagne, J., Rouleau, M., Poirier, G. (2012), PARP-1 Activation— Bringing the Pieces Together, Science, 336:678-279.
Gehrke, T., Lischke, U., Gasteiger, K., Schneider, S., Arnold, S., Muller, H., Stephenson, D., Zipse, H., Carell, T. (2013), Unexpected non-Hoogsteen–based mutagenicity mechanism of FaPy-DNA lesions, Nat Chem Biol, 9:455-461.
Greenberg, M. (2012), Purine Lesions Formed in Competition With 8-Oxopurines From Oxidative Stress, Acc Chem Res, 45:588-597.
Georgakilas, A., P. O'Neill, and R. Stewart. (2013), “Induction and repair of clustered DNA lesions: What do we know so far?”, Radiation Research, Vol.180/1, Radiation Research Society, Indianapolis, https://doi.org/10.1667/RR3041.1.
Hashimoto, K., Tominaga, Y., Nakabeppu, Y., Moriya, M. (2004), Futile short-patch DNA base excision repair of adenine:8-oxoguanine mispair, Nucleic Acids Res, 32:5928-5934.
Hegde, M. L. et al. (2012). “Oxidized base damage and single-strand break repair in mammalian genomes: role of disordered regions and posttranslational modifications in early enzymes”, Progress in molecular biology and translational science, 110, 123–153, https://doi.org/10.1016/B978-0-12-387665-2.00006-7
Kozbenko, T. et al. (2022), “Deploying elements of scoping review methods for adverse outcome pathway development: a space travel case example”, International Journal of Radiation Biology, 1–12. https://doi.org/10.1080/09553002.2022.2110306
Le Page, F., Klunglund, A., Barnes, D., Sarasin, A., Boiteux, S. (2000), Transcription coupled repair of 8-oxoguanine in murine cells: The Ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences, Proc Natl Acad Sci USA, 97:8397-8402.
Li, Y., Song, M., Kasai, H., Kawai, K. (2013), Generation and threshold level of 8-OHdG as oxidative DNA damage elicited by low dose ionizing radiation, Genes Environ, 35:88-92.
Ma, H., Wang, J., Abdel-Rahman, S., Boor, P., Firoze, M. (2008), Oxidative DNA damage and its repair in rat spleen following subchronic exposure to aniline, Toxicol Appl Pharmacol, 233:247-253.
Maddukuri, L., Ketkar, A., Eddy, S., Zafar, M., Eoff, R. (2014), The Werner syndrome protein limits the error-prone 8-oxo-dG lesion bypass activity of human DNA polymerase kappa, Nucleic Acids Res, 42:12027-12040.
Markkanen, E. (2017), Not breathing is not an option: How to deal with oxidative DNA damage, DNA Repair, 59:82-105.
Markkanen, E., Castrec, B., Vilani, G., Hubscher, U. (2012), A switch between DNA polymerases δ and λ promotes error-free bypass of 8-oxo-G lesions, Proc Natl Acad Sci USA, 27:931-940.
Mesa, R. and S. Bassnett. (2013), “UV-B-induced DNA damage and repair in the mouse lens”, Investigative Ophthalmology and Visual Science, Vol.54/10, Association for Research in Vision and Ophthalmology, Rockville, https://doi.org/10.1167/iovs.13-12644.
Pastoriza-Gallego, M., Armier, J., Sarasin, A. (2007), Transcription through 8-oxoguanine in DNA repair-proficient and Csb−/Ogg1− DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context, Mutagenesis, 22:343-351.
Pearson, C., Shikazono, N., Thacker, J., O’Neill, P. (2004), Enhanced mutagenic potential of 8-oxo-7,8-dihydroguanine when present within a clustered DNA damage site, Nucleic Acids Res, 32:263-270.
Poetsch, A. (2020), “The genomics of oxidative DNA damage, repair, and resulting mutagenesis”, Computational and Structural Biotechnology Journal, Vol.18, Elsevier, Amsterdam, https://doi.org/10.1016/j.csbj.2019.12.013.
Seager, A., Shah, U., Mikhail, J., Nelson, B., Marquis, B., Doak, S., Johnson, G., Griffiths, S., Carmichael, P., Scott, S., Scott, A., Jenkins, G. (2012), Pro-oxidant Induced DNA Damage in Human Lymphoblastoid Cells: Homeostatic Mechanisms of Genotoxic Tolerance, Toxicol Sci, 128:387-397.
Shafirovich, V., Kropachev, K., Anderson, T., Li, Z., Kolbanovskiy, M., Martin, B., Sugden, K., Shim, Y., Min, J., Ceacintov, N. (2016), Base and Nucleotide Excision Repair of Oxidatively Generated Guanine Lesions in DNA, J Biol Chem, 291:5309-5319.
Shah, A., Gray, K., Figg, N., Finigan, A., Starks, L., Bennett, M. (2018), . Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis, Circulation, 138:1446-1462.
Sharma, V., Collins, L., Chen, T., Herr, N., Takeda, S., Sun, W., Swenberg, J., Nakamura, J. (2016), Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations, Oncotarget, 7:25377-25390.
Sokhansanj, B., Wilson III, D. (2004), Oxidative DNA damage background estimated by a system model of base excision repair, Free Rad Biol Med, 37:433-427.
Swenberg, J., Lu, K., Moeller, B., Gao, L., Upton, P., Nakamura, J., Starr, T. (2011), Endogenous versus Exogenous DNA Adducts: Their Role in Carcinogenesis, Epidemiology, and Risk Assessment, Toxicol Sci, 120:S130-S145.
Taggart, D., Fredrickson, S., Gadkari, V., Suo, Z. (2014), Mutagenic Potential of 8-Oxo-7,8-dihydro-2′-deoxyguanosine Bypass Catalyzed by Human Y-Family DNA Polymerases, Chem Res Toxicol, 27:931-940.
Wang, C., Yang, J., Lu, D., Fan, Y., Zhao, M., Li, Z. (2016), Oxidative stress-related DNA damage and homologous recombination repairing induced by N,N-dimethylformamide , J Appl Toxicol, 36:936-945.
Wang, R., Li, C., Qiao, P., Xue, Y., Zheng, X., Chen, H., Zeng, X., Liu, W., Boldogh, I., Ba, X. (2018), OGG1-initiated base excision repair exacerbates oxidative stress-induced parthanatos, Cell Death and Disease, 9:628.
Yang, N., Galick, H., Wallace, S. (2004), Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks, DNA Repair, 3:1323-1334.
Yoshikawa, Y., Yamasaki, A., Takatori., K., Suzuki, M., Kobayashi, J., Takao, M., Zhang-Akiyama, Q. (2015), Excess processing of oxidative damaged bases causes hypersensitivity to oxidative stress and low dose rate irradiation, Free Radic Res, 49:1239-1248.
Relationship: 1910: Inadequate DNA repair leads to Increase, DNA strand breaks
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Oxidative DNA damage leading to chromosomal aberrations and mutations | adjacent | High | Low |
| Alkylation of DNA leading to reduced sperm count | adjacent | ||
| Excessive reactive oxygen species leading to growth inhibition via oxidative DNA damage | adjacent | ||
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell cycle disruption | adjacent | High | Low |
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell death | adjacent | High | Moderate |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages |
| Sex | Evidence |
|---|---|
| Unspecific |
This KER applies to any cell type that has DNA repair capabilities.
Key Event Relationship Description
Inadequate repair of DNA damage includes incorrect repair (i.e., incorrect base insertion), incomplete repair (i.e., accumulation of repair intermediates such as strand breaks, stalled replications forks, and/or abasic sites), and absent repair resulting in the retention of DNA damage.
It is well-established that DNA excision repair pathways require DNA strand breakage for removing the damaged sites; for example, base excision repair (BER) of oxidative lesions involves removal of oxidized bases by glycosylases followed by cleavage of the DNA strand 5’ from the abasic site. If the repair process is disrupted at this point, repair intermediates including single strand breaks (SSB) may persist in the DNA. A SSB can turn into a double strand break (DSB) if it occurs sufficiently close to another SSB on the opposite strand. SSBs can be converted into DSBs when helicase unwinds the DNA strands during replication. Furthermore, SSBs and abasic sites can act as replication blocks causing the replication fork to stall and collapse, giving rise to DSBs (Minko et al., 2016; Whitaker et al., 2017).
The two most common DSB repair mechanisms are non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ is may favoured over HR and has also been shown to be 104 times more efficient than HR in repairing DSBs (Godwin et al., 1994; Benjamin and Little, 1992). There are two subtypes of NHEJ: canonical NHEJ (C‐NHEJ) or alternative non-homologous end joining (alt-NHEJ). During C-NHEJ, broken ends of DNA are simply ligated together. In alt‐NHEJ, one strand of the DNA on either side of the break is resected to repair the lesion (Betermeir et al., 2014). Although both repair mechanisms are error‐prone (Thurtle‐Schmidt and Lo, 2018), alt-NHEJ is considered more error-prone than C-NHEJ (Guirouil-Barbat et al., 2007; Simsek and Jasin, 2010). While NHEJ may prevent cell death due to the cytotoxicity of DSBs, it may lead to mutations and genomic instability downstream.
Evidence Supporting this KER
Biological Plausibility1. DNA strand breaks generated due to faulty attempted repair
Excision repair pathways require the induction of SSB as part of damage processing. Increases in DNA lesions may lead to the accumulation of intermediate SSB. Attempted excision repair of lesions on opposite strands can turn into DSBs if the two are in close proximity (Eccles et al., 2010). Generation of DSBs has been observed in both nucleotide excision repair (NER) and BER (Ma et al., 2009; Wakasugi et al., 2014).
Previous studies have demonstrated that an imbalance in one of the multiple steps of BER can lead to an accumulation of repair intermediates and failed repair. It is highly likely that a disproportionate increase in oxidative DNA lesions compared to the level of available BER glycosylases leads to an imbalance between lesions and the initiating step of BER (Brenerman et al., 2014). Accumulation of oxidative lesions, abasic sites, and SSBs generated from OGG1, NTH1, and APE1 activities would be observed as a result. Moreover, studies have reported accumulation of SSB due to OGG1- and NHT1-overexpression (Yang et al., 2004; Yoshikawa et al., 2015; Wang et al., 2018). BER repair intermediates have been observed to interfere with transcription as well (Kitsera et al., 2011). While overexpression may lead to imbalanced lyase activities that generate excessive SSB intermediates, deficiency of these enzymes is also known to cause an accumulation of oxidative lesions that could lead to strand breaks downstream. Hence, both the overexpression and deficiencies of repair enzymes can lead to strand breaks due to excessive activity or inadequate repair, respectively.
2. DNA strand breaks generated due to replication stress caused by accumulated DNA lesions
Retention of DNA lesions (i.e., damaged bases and SSB) can interfere with the progression of the replication fork. Thymidine glycol is an example of an oxidative DNA lesion that acts as a replication block (Dolinnaya et al., 2013). Persistent replication fork stalling and dissociation of replication machinery are known to cause the replication fork to collapse, which generates highly toxic DSBs (Zeman and Cimprich, 2014; Alexander and Orr-Weaver, 2016). Fork stalling also increases the risk of two replication forks colliding with each other, generating DSBs.
In addition, the replication fork can collide with SSBs generated during BER, hindering the completion of repair and giving rise to DSBs (Ensminger et al., 2014).
In vitro studies with empirical evidence are shown below for select DNA repair pathways. These studies build in elements of essentiality (modulation of DNA repair), as well as dose and incidence concordance. The primary evidence is essentiality, where repair is genetically modulated in some way. Because multiple lines of evidence are considered within individual studies, we present the data by source of evidence (in vitro versus in vivo) rather than by type of empirical evidence (dose, incidence, or temporal concordance; essentiality) to avoid repetitive use of the same studies.
Inadequate repair of oxidative lesions
- Concentration concordance of strand breaks in repair-deficient and –proficient cells (insufficient repair) (Wu et al., 2008)
- In a study using A549 human adenocarcinoma cells, DNA strand breaks in hOGG1-proficient and hOGG1-deficient cells were compared following exposure to increasing concentrations of bleomycin.
- Strand breaks were measured as DNA migration length in alkaline comet assay after 3 hours of exposure to six increasing concentrations (0.05, 0.25, 0.5, 1, 5, and 10 mg/L).
- Concentration-dependent increase in strand breaks was observed in both cell types; however, at all concentrations significantly more strand breaks (p<0.05) were present in the hOGG1-deficient cells than in the proficient cells, demonstrating insufficient repair of oxidative lesions leading to DNA strand breaks.
- Thus, this evidence supports the essentiality of inadequate DNA repair as a modulator of the downstream KE.
- Incomplete OGG1-initiated base excision repair (BER) leads to DNA strand breaks (Wang et al., 2018):
- In a study using mouse embryonic fibroblasts (MEF), Ogg1+/+ and Ogg1-/- cells were treated with increasing concentrations of H2O2 for varying durations
Higher levels of 8-oxodG were detected in Ogg1-/- cells compared to Ogg1+/+ cells after treatment with 400 µM H2O2 at all time points (5, 15, 30, 60, and 90 min)- Demonstrates insufficient removal of 8-oxo-dG in OGG1-deficient cells
- Significantly more strand breaks, as indicated by the higher % of TUNEL-positive cells (p<0.001), were detected in Ogg1+/+ cells compared to Ogg1-/- cells after exposure to 400 µM H2O2 for 3 hours
- Both cell types showed a very similar increase in DNA strand breaks at lower concentrations (50, 100, and 200
µM) and there was no significant difference between Ogg1+/+ and Ogg1-/- cells at these concentrations – this suggests that up to a certain level of oxidative damage, OGG1-initiated BER does not exacerbate strand breaks but when oxidative stress is excessive (at 400µM in this study), OGG1-initiated BER is compromised and leads to increased strand breaks (incomplete repair)
- Both cell types showed a very similar increase in DNA strand breaks at lower concentrations (50, 100, and 200
- Finally, DNA strand breaks in both cell types were measured using both alkaline and neutral comet assay after a 30- minute exposure to 400µM H2O2; while there was an increase in the olive tail moment (indicating DNA strand breaks) in both cell types compared to the control, the increase of strand breaks in Ogg1+/+ cells was significantly larger than in Ogg1-/- cells in both assays (p<0.001)
- In a study using mouse embryonic fibroblasts (MEF), Ogg1+/+ and Ogg1-/- cells were treated with increasing concentrations of H2O2 for varying durations
Inadequate repair of alkylated DNA
- Interference of N-methylpurine DNA glycosylase (MPG)-initiated BER by replication leading to strand breaks (Ensminger et al., 2014)
- A549 human alveolar basal epithelial cells were exposed to increasing concentrations of methylmethane sulfonate (MMS) for 1 hour and replicating cells were labeled using a thymidine analogue, 5-ethynyl-2’-desoxyuridine (EdU).
- In S-phase cells, MMS concentration-dependent increase in γH2AX foci was detected (70 foci/cell at the highest concentration). In contrast, γH2AX foci were not detected G1- and G2-phase cells until the highest concentration (15 foci/cell).
- MPG-depleted cells in S-phase showed no significant increase in γH2AX foci, while the control cells showed significant MMS concentration-dependent increases.
- These results suggest interference of MPG-initiated BER by replication, leading to DSBs, and that the depletion of MPG decreases the probability of strand breaks in S-phase (evidence of essentiality of ‘inadequate repair’ to KEdown).
Inadequate mismatch repair
- Incomplete/incorrect mismatch repair (MMR) leads to DNA strand breaks (Peterson-Roth et al., 2005):
- MLH1 (MMR protein)-deficient and -proficient HCT116 human colon cancer cells were treated with 30µM K2CrO4 (DNA crosslinking, Cr adducts, protein-DNA crosslinking, DNA oxidation) for 3, 6, and 12 hours and γH2AX foci (biomarker of DNA DSB) were scored by fluorescence microscopy
- At 6 and 12 hours, MLH1+ cells had higher percentage of γH2AX foci than MLH1- cells
- The futile repair model of MMR suggests that strand breaks arise from MMR attempting repeatedly to repair the newly synthesized strand opposite adducts in S and G2 phases; approximately 80% of the γH2AX-positive MLH1+ cells were in G2 phase 12 hours after a 3-hour exposure to 20 µM Cr(VI), while the level was five times lower in MLH1- cells, suggesting that the MMR-induced DSB occurred following DNA synthesis; this supports the futile repair model and demonstrates inadequate repair
Inadequate Repair of DSBs
- Rydberg et al. [2005] exposed GM38 primary human dermal fibroblasts to increasing doses of linear electron transfer (LET) radiation of helium and iron ions (Rydberg et al., 2005).
- The cells were allowed to recover for 16 hours following irradiation.
- Unrepaired DSBs were measured after recovery using PFGE.
- There was a dose-dependent increase in unrepaired DSBs due to both ion exposures.
- Increase in persistent unrepaired DSBs with increasing dosage indicates exceeded repair capacity.
- DSB repair was also monitored by measuring γH2AX foci 0.05 - 24 hours after irradiation.
- DSBs decreased over time and less than 1 foci per cell on average remained in MRC-5 cells 24hours after 0.02, 0.2 and 2 Gy exposures.
- Repair was slower in 180BR cells, particularly for the 2 Gy exposure, where 20 foci per cell remained after 24 h.
- A follow-up study by the same group, found similar results for MRC-5 and 180BR cells exposed to 0.02 and 0.2 Gy of X-rays (Kühne et al., 2004).
- Rothkamm and Löbrich (2003) exposed MRC-5 primary human lung fibroblasts (repair-proficient) and 180BR DNA ligase IV-deficient human fibroblasts to 10 and 80 Gy of X-rays (Rothkamm and Lobrich, 2003).
- DNA ligase IV deficiency results in impaired NHEJ
- DSB repair was monitored using PFGE by measuring the % of DSBs remaining after 0.25, 2, and 24 h following irradiation.
- DSBs decreased over time and, eventually, less than 10% of the DSBs remained in MRC-5 cells after 24h following both 80 and 10 Gy exposures.
- Repair was noticeably slower in 180BR cells, where the clearance of DSBs was hindered and approximately 40 and 20% of the DSBs remained at 24 hours following 80 and 10 Gy exposures, respectively.
- The above demonstrates defective DNA repair leading to persistent DSBs.
Uncertainties and Inconsistencies
- A variety of confounding factors and genetic characteristics (i.e., SNPs) may modulate which repair pathways are invoked and the degree to which they are inadequate. These have yet to be fully defined.
- Both protective and damaging effects of OGG1 against strand breaks have been described in the literature. As demonstrated in the section above, the effect of OGG1-deficiency (BER-initiating enzyme) is observed to be different in different cell types; Wang et al. (2018) demonstrated strand breaks exacerbated by excessive OGG1 activity, while Wu et al. (2008) and Shah et al. (2018) demonstrated increased strand breaks due to lack of repair in mammalian cells in culture (Shah et al., 2018; Wu et al., 2008; Wang et al., 2018). Cell cycle and replication may influence the effect of DNA repair on exacerbating strand breaks.
- Dahle et al. (2008) exposed wild type and OGG1-overexpressing Chinese hamster ovary cells, AS52, to UVA. While OGG1-overexpression prevented the accumulation of Fpg-sensitive lesions (e.g., 8-oxo-dG and FaPyG) that were observed in wild type cells 4 hours after irradiation, there was no difference in the amount of strand breaks in the two cell types at 4h (Dahle et al., 2008).
- A recent study suggests that the NHEJ may be more accurate than previously thought (reviewed in Betermier et al., 2014). The accuracy of NHEJ may be dependent on the structure of the termini. The termini processing rather than the NHEJ itself is thus argued to be error-prone process (Betemier et al., 2014).
References
Alexander, J., Orr-Weaver, T. (2016), Replication fork instability and the consequences of fork collisions from rereplication, Genes Dev, 30:2241-2252.
Brenerman, B., Illuzzi, J., Wilson III, D. (2014), Base excision repair capacity in informing healthspan, Carcinogenesis, 35:2643-2652.
Dahle, J., Brunborg, G., Svendsrud, D., Stokke, T., Kvam, E. (2008), Overexpression of human OGG1 in mammalian cells decreases ultraviolet A induced mutagenesis, Cancer Lett, 267:18-25.
Dolinnaya, N., Kubareva, E., Romanova, E., Trikin, R., Oretskaya, T. (2013), Thymidine glycol: the effect on DNA molecular structure and enzymatic processing, Biochimie, 95:134-147.
Eccles, L., Lomax, M., O’Neill, P. (2010), Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage, Nucleic Acids Res, 38:1123-1134.
Ensminger, M., Iloff, L., Ebel, C., Nikolova, T., Kaina, B., Lobrich, M. (2014), DNA breaks and chromosomal aberrations arise when replication meets base excision repair, J Cell Biol, 206:29.
Kitsera, N., Stathis, D., Luhnsdorf, B., Muller, H., Carell, T., Epe, B., Khobta, A. (2011), 8-Oxo-7,8-dihydroguanine in DNA does not constitute a barrier to transcription, but is converted into transcription-blocking damage by OGG1, Nucleic Acids Res, 38:5926-5934.
Kühne, M., E. Riballo, N. Rief, K. Rothkamm, P. Jeggo, & M. Löbrich (2004), "A Double-Strand Break Repair Defect in ATM-Deficient Cells Contributes to Radiosensitivity", Cancer Res, 64(2): 500-508.
Ma, W., Panduri, V., Sterling, J., Van Houten, B., Gordenin, D., Resnick, M. (2009), The Transition of Closely Opposed Lesions to Double-Strand Breaks during Long-Patch Base Excision Repair Is Prevented by the Coordinated Action of DNA Polymerase and Rad27/Fen1 , Mol Cell Biol, 29:1212-1221.
Minko, I., Jacobs, A., de Leon, A., Gruppi, F., Donley, N., Harris, T., Rizzo, C., McCullough, A., Lloyd, R.S. (2016), Catalysts of DNA Strand Cleavage at Apurinic/Apyrimidinic Sites, Sci Rep, 6.
Peterson-Roth, E., Reynolds, M., Quievryn, G., Zhitkovich, A. (2005), Mismatch Repair Proteins Are Activators of Toxic Responses to Chromium-DNA Damage, Mol Cell Biol, 25:3596-3607.
Rothkamm, K., Lobrich, M. (2003), Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses, Proc Natl Acad Sci USA, 100:5057-5062.
Rydberg, B., Cooper, B., Cooper, P., Holley, W., Chatterjee, A. (2005), Dose-Dependent Misrejoining of Radiation-Induced DNA Double-Strand Breaks in Human Fibroblasts: Experimental and Theoretical Study for High- and Low-LET Radiation, Radiat Res, 163:526-534.
Shah, A., Gray, K., Figg, N., Finigan, A., Starks, L., Bennett, M. (2018), . Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis, Circulation, 138:1446-1462.
Wakasugi, M., Sasaki, T., Matsumoto, M., Nagaoka, M., Inoue, K., Inobe, M., Horibata, K., Tanaka, K., Matsunaga, T. (2014), Nucleotide Excision Repair-dependent DNA Double-strand Break Formation and ATM Signaling Activation in Mammalian Quiescent Cells, J Biol Chem, 289:28730-28737.
Wang, R., Li, C., Qiao, P., Xue, Y., Zheng, X., Chen, H., Zeng, X., Liu, W., Boldogh, I., Ba, X. (2018), OGG1-initiated base excision repair exacerbates oxidative stress-induced parthanatos, Cell Death and Disease, 9:628.
Whitaker, A., Schaich, M., Smith, M.S., Flynn, T., Freudenthal, B. (2017), Base excision repair of oxidative DNA damage: from mechanism to disease, Front Biosci, 22:1493-1522.
Wu, M., Zhang, Z., Che, W. (2008), Suppression of a DNA base excision repair gene, hOGG1, increases bleomycin sensitivity of human lung cancer cell line, Toxicol App Pharmacol, 228:395-402.
Yang, N., Galick, H., Wallace, S. (2004), Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks, DNA Repair, 3:1323-1334.
Yoshikawa, Y., Yamasaki, A., Takatori., K., Suzuki, M., Kobayashi, J., Takao, M., Zhang-Akiyama, Q. (2015), Excess processing of oxidative damaged bases causes hypersensitivity to oxidative stress and low dose rate irradiation, Free Radic Res, 49:1239-1248.
Zeman, M., Cimprich, K. (2014), Causes and Consequences of Replication Stress, Nat Cell Biol, 12:2-9.
Relationship: 3480: Increase, DNA strand breaks leads to Cell cycle disruption
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Excessive reactive oxygen species leading to growth inhibition via oxidative DNA damage | adjacent | ||
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell cycle disruption | adjacent | High | Moderate |
Relationship: 3363: Cell cycle disruption leads to Decrease, Cell proliferation
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| DBDPE-induced DNA damage increase in liver leading to Non-alcoholic fatty liver disease via liver steatosis and inhibition of regeneration | adjacent | Not Specified | Not Specified |
| Excessive reactive oxygen species leading to growth inhibition via oxidative DNA damage | adjacent | ||
| Excessive reactive oxygen species leading to growth inhibition via protein oxidation and cell cycle disruption | adjacent | ||
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell cycle disruption | adjacent | High | Moderate |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages | Moderate |
| Sex | Evidence |
|---|---|
| Unspecific | Moderate |
This KER is applicable to proliferating eukaryotic cells and tissues in which cell-cycle progression is required for cell number increase. It is broadly relevant across algae, invertebrates, fish, mammals and human-derived cell systems when the cells or tissues under study are actively proliferating or can be stimulated to proliferate. Applicability is strongest for developmental, regenerative, immune, epithelial, tumor, algal growth or cell culture contexts, where decreased cell proliferation is readily measured.
The KER is less directly applicable to terminally differentiated non-dividing cells or tissues in which proliferation is not a meaningful endpoint. It should also be interpreted carefully when decreased proliferation is inferred from metabolic viability assays alone, because changes in ATP, mitochondrial activity or cytotoxicity may confound proliferation measurements. Species, sex and life stage are best viewed as modifiers of sensitivity rather than determinants of whether the relationship can occur.
Key Event Relationship Description
This KER describes the causal and predictive relationship by which disruption of the cell cycle leads to decreased cell proliferation. Cell proliferation requires cells to progress through an ordered series of phases, including G1, S, G2 and M phase, with checkpoints that monitor cell size, DNA replication, DNA damage, spindle assembly and other conditions necessary for successful division. When these processes are disrupted, cells may arrest at a checkpoint, delay progression, fail to replicate DNA, fail mitosis, enter senescence, or undergo cell death. Any of these outcomes reduces the fraction of cells completing successful division and therefore decreases the net rate of cell proliferation (Hartwell and Weinert, 1989; Nurse, 2000; Malumbres and Barbacid, 2009).
The upstream KE is deliberately defined broadly as "cell cycle, disrupted" to preserve modularity across AOPs. It can include G0/G1 arrest, S-phase arrest, G2/M delay, mitotic arrest, checkpoint activation, failure of DNA synthesis, altered cyclin/CDK regulation, spindle checkpoint disruption or permanent cell-cycle exit. The downstream KE, "Decrease, Cell proliferation", refers to a reduction in the rate of increase in cell number or proliferative capacity, measured by cell counts, DNA synthesis, EdU/BrdU incorporation, Ki-67 or PCNA markers, colony formation, growth rate of unicellular organisms, or similar proliferation endpoints.
Evidence Supporting this KER
Biological PlausibilityBiological plausibility of this KER is high. Cell proliferation requires successful completion of the cell cycle. Checkpoints that delay or block progression are conserved control mechanisms that prevent cells from dividing when DNA is damaged, DNA replication is incomplete, chromosomes are not correctly attached to the spindle, or other cellular conditions are incompatible with successful division (Hartwell and Weinert, 1989; O'Connell et al., 2000; Nurse, 2000). If disruption persists, cells fail to complete mitosis, enter senescence, or activate cell death pathways, resulting in reduced net cell accumulation. The AOP-Wiki Event 1505 page similarly describes cell-cycle disruption as a disruption of G1, S, G2, M or G0 progression that can lead to decreased cell number (AOP-Wiki, 2026b).
The structural and functional relationship between the KEs is direct: the upstream KE alters the process required to generate daughter cells, while the downstream KE represents the measurable decrease in cell proliferation. Cyclins, cyclin-dependent kinases, checkpoint kinases, p53/p21 signaling, DNA damage response pathways and spindle checkpoint mechanisms all provide mechanistic links through which upstream cell-cycle disruption can reduce proliferation (Malumbres and Barbacid, 2009; Cuddihy and O'Connell, 2003).
Empirical EvidenceEmpirical support for this KER is moderate to high. The relationship is supported by many studies across cell biology and toxicology showing that stressor-induced cell-cycle arrest or disruption is accompanied by decreased cell number, colony formation, DNA synthesis or population growth. Within the ROS-growth evidence set, the most relevant evidence comes from systems in which genotoxic or oxidative stress is associated with cell-cycle responses and reduced proliferation or cell accumulation. However, not every study measures the two KEs in the same time course, so the empirical call is not uniformly high across all taxa and stressor classes.
Empirical Evidence Table
|
Biological system |
Stressor or perturbation |
Evidence relevant to KER |
Interpretation |
|
Green algae |
DNA-damaging agents such as zeocin or N-OH-2-AAF |
DNA damage-induced changes in cell-cycle progression and division outcomes were reported in green algal systems, demonstrating that damage-associated cell-cycle responses can alter cell division behavior (David et al., 2009; Hlavová et al., 2011). |
Supports relevance of cell-cycle disruption to algal cell division/proliferation, although responses can be species- and phase-specific. |
|
Green algae, Chlamydomonas reinhardtii |
Isoproturon and cadmium |
The combined exposure reduced chlorophyll and photosynthetic performance, increased oxidative damage markers, and reduced algal fitness endpoints; this provides supporting context for stressor-induced disruption of growth-related cellular processes in algae (Qiu et al., 2024). |
Supportive but indirect for this specific KER; strongest for stressor effects on growth and oxidative damage rather than a direct cell-cycle-to-proliferation linkage. |
|
Embryonic zebrafish cells |
Silver nanoparticles and ionic silver |
Silver exposure triggered DNA damage/repair responses in embryonic zebrafish cells, with endpoints relevant to damage response and cell-cycle control (Quevedo et al., 2021). |
Supports relevance of the DNA damage response/cell-cycle context in fish cell systems. |
|
Human Jurkat T cells |
Silver nanoparticles |
AgNP exposure activated stress signaling and induced DNA damage, cell-cycle arrest and apoptosis; the study also reported cytotoxicity/viability effects (Eom and Choi, 2010). |
Supports co-occurrence of cell-cycle arrest with impaired cell survival/proliferation in a human cell model. |
|
Human and mammalian cells |
Genotoxic and cell-cycle regulatory perturbations |
Large mechanistic literature shows that checkpoint activation, CDK inhibition and cell-cycle arrest reduce DNA synthesis, mitotic entry and cell accumulation (Hartwell and Weinert, 1989; Cuddihy and O'Connell, 2003; Malumbres and Barbacid, 2009). |
Provides broad empirical and mechanistic support for the general KER. |
Uncertainties and Inconsistencies
The main uncertainty is that cell-cycle disruption is a broad upstream KE and can represent different biological states. Transient checkpoint activation may delay proliferation without causing a sustained decrease in cell number, whereas persistent arrest, mitotic failure or permanent cell-cycle exit has a stronger effect on proliferation. The magnitude of the downstream response therefore depends on the duration and reversibility of the upstream cell-cycle perturbation.
Another uncertainty is that decreased cell proliferation can be measured by multiple endpoints, including cell counts, DNA synthesis, colony formation or metabolic activity. Some assays, such as MTT or resazurin, may reflect both proliferation and viability/metabolic state, which can complicate interpretation. Additionally, cell-cycle disruption may lead to cell death in some contexts rather than simply decreased proliferation, so the downstream response may diverge depending on severity and cell type. In algal and early developmental systems, reduced population growth can result from both proliferation effects and other processes such as photosynthetic inhibition, energy depletion or cell death.
Quantitative Understanding of the Linkage
Quantitative understanding of this KER is moderate. The qualitative relationship is well established: cells that cannot progress through the cell cycle cannot complete division, and sustained arrest therefore reduces proliferation. However, a general quantitative model that predicts the magnitude of decreased proliferation from a given measure of cell-cycle disruption has not been established across species, cell types, stressors and assay platforms.
Response-response relationshipThe relationship can be quantified in specific experimental systems by relating the fraction of cells in each cell-cycle phase, the proportion of cells positive for DNA synthesis markers such as BrdU or EdU, mitotic index, checkpoint marker intensity, or duration of arrest to subsequent changes in cell number, population doubling time or colony-forming ability. The time scale varies from hours for checkpoint activation and DNA synthesis inhibition to days for measurable reductions in population growth or colony formation. The relationship is expected to be nonlinear: a transient delay may produce limited or reversible effects, whereas persistent arrest or permanent cell-cycle exit can sharply reduce proliferation.
Quantitative prediction is complicated by several factors, including baseline growth rate, synchronization state of the cell population, cell-cycle phase at exposure, checkpoint competence, repair capacity, cell death, and assay choice. Therefore, quantitative interpretation should be made within a defined biological system and ideally with paired upstream and downstream measurements collected across multiple time points and exposure concentrations.
Known modulating factors|
Modulating factor |
Details |
Influence on KER |
Supporting evidence |
|
Cell type and proliferative state |
Rapidly dividing, quiescent, differentiated, stem-like or senescent cells. |
Rapidly dividing cells are more sensitive to cell-cycle disruption; quiescent or terminally differentiated cells may show little proliferation effect. |
Nurse, 2000; Malumbres and Barbacid, 2009. |
|
Cell-cycle phase at exposure |
G1, S, G2 or M phase when the stressor occurs. |
Phase determines whether disruption blocks DNA synthesis, mitotic entry, mitosis, or return to cycle; this affects time course and magnitude of proliferation decrease. |
Hartwell and Weinert, 1989; Hlavová et al., 2011. |
|
DNA damage response and checkpoint capacity |
p53, p21, ATM/ATR, Chk1/Chk2 and related checkpoint pathways. |
Strong checkpoint activation can increase arrest and decrease proliferation; impaired checkpoints may permit division with damage or shift outcome toward cell death. |
O'Connell et al., 2000; Cuddihy and O'Connell, 2003. |
|
Repair capacity and stressor severity |
Extent and persistence of DNA damage, oxidative stress or spindle disturbance. |
Mild, reversible disruption may delay proliferation; severe or persistent disruption can cause permanent arrest, senescence or cell death. |
Cuddihy and O'Connell, 2003; Eom and Choi, 2010. |
|
Compensatory growth and recovery |
Post-exposure recovery, tissue repair and regenerative proliferation. |
Recovery mechanisms can reduce the apparent downstream effect on proliferation if the upstream disruption is transient. |
Malumbres and Barbacid, 2009. |
References
AOP-Wiki. 2026a. Relationship 3363: Cell cycle, disrupted leads to Decrease, Cell proliferation. AOP-Wiki. Accessed 14 May 2026.
AOP-Wiki. 2026b. Event 1505: Cell cycle, disrupted. AOP-Wiki. Accessed 14 May 2026.
Cuddihy AR, O'Connell MJ. 2003. Cell-cycle responses to DNA damage in G2. International Review of Cytology 222:99-140. https://doi.org/10.1016/S0074-7696(02)22013-6.
Eom HJ, Choi J. 2010. p38 MAPK activation, DNA damage, cell cycle arrest and apoptosis as mechanisms of toxicity of silver nanoparticles in Jurkat T cells. Environmental Science & Technology 44(21):8337-8342. https://doi.org/10.1021/es1020668.
Hartwell LH, Weinert TA. 1989. Checkpoints: controls that ensure the order of cell cycle events. Science 246(4930):629-634. https://doi.org/10.1126/science.2683079.
Hlavová M, Čížková M, Vítová M, Bišová K, Zachleder V. 2011. DNA damage during G2 phase does not affect cell cycle progression of the green alga Scenedesmus quadricauda. PLoS ONE 6(5):e19626. https://doi.org/10.1371/journal.pone.0019626.
Malumbres M, Barbacid M. 2009. Cell cycle, CDKs and cancer: a changing paradigm. Nature Reviews Cancer 9(3):153-166. https://doi.org/10.1038/nrc2602.
Nurse P. 2000. A long twentieth century of the cell cycle and beyond. Cell 100(1):71-78. https://doi.org/10.1016/S0092-8674(00)81684-0.
O'Connell MJ, Walworth NC, Carr AM. 2000. The G2-phase DNA-damage checkpoint. Trends in Cell Biology 10(7):296-303. https://doi.org/10.1016/S0962-8924(00)01773-6.
Qiu CB, Tang J, Chen G, Yang H, Liu J. 2024. Single and joint bioaccumulation and toxicity of isoproturon and cadmium in green algae (Chlamydomonas reinhardtii). Chemical and Biological Technologies in Agriculture 11:97. https://doi.org/10.1186/s40538-024-00628-3.
Quevedo AC, Lynch I, Valsami-Jones E. 2021. Cellular repair mechanisms triggered by exposure to silver nanoparticles and ionic silver in embryonic zebrafish cells. Environmental Science: Nano 8(9):2507-2522. https://doi.org/10.1039/D1EN00422K.
Relationship: 2205: Decrease, Cell proliferation leads to Decrease, Growth
AOPs Referencing Relationship
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| zebrafish | Danio rerio | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Embryo | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
Taxonomic applicability
Relationship 2205 is considered applicable to all eukaryotes (both unicellular and multicellular), as growth (or population growth of alga) is well known to be achieved through cell proliferation in animals, plants and some microorganisms.
Sex applicability
Relationship 2205 is considered applicable to both all sexes, as cell proliferation leading to growth is a fundamental process and not sex-specific.
Life-stage applicability
Relationship 2205 is considered applicable to all life stages, as cell proliferation leading to growth is essential for maintaining basic biological processes throughout an organism’s life.
Key Event Relationship Description
This key event relationship describes reduced cell proliferation (cell growth, division or a combination of these) leading to reduced tissue, organ or individual growth.
Evidence Supporting this KER
The overall evidence supporting Relationship 2205 is considered moderate.
Biological PlausibilityThe biological plausibility of Relationship 2205 is considered high.
Rationale: The biological structural and functional relationship between cell proliferation and growth is well established. It is commonly accepted that the size of an organism, organ or tissue is dependent on the total number and volume of the cells it contains, and the amount of extracellular matrix and fluids (Conlon 1999). Impairment to cell proliferation can logically affect tissue and organismal growth.
Empirical EvidenceThe empirical support of Relationship 2205 is considered low.
Rationale: Because cell proliferation is typically measured in vitro, while growth of an organism is measured in vivo, few studies have measured both in the same experiment. There is one zebrafish study reporting concordant relationship between reduced cell proliferation and embryo growth with some inconsistencies (Bestman 2015).
Uncertainties and Inconsistencies- In zebrafish embryos exposed to 2,4-DNP, significant growth inhibition (AO), as indicated by whole embryo length, caudal primary (CaP) motor neuron axons and otic vesicle length (OVL) ratio after 21h, somite width and eye diameter after 45h exposure was identified, after 21h, whereas a non- significant reduction in cell proliferation was observed (Bestman 2015).
References
Bestman JE, Stackley KD, Rahn JJ, Williamson TJ, Chan SS. 2015. The cellular and molecular progression of mitochondrial dysfunction induced by 2,4-dinitrophenol in developing zebrafish embryos. Differentiation 89:51-69. DOI: 10.1016/j.diff.2015.01.001.
Binder BJ, Landman KA, Simpson MJ, Mariani M, Newgreen DF. 2008. Modeling proliferative tissue growth: a general approach and an avian case study. Phys Rev E Stat Nonlin Soft Matter Phys 78:031912. DOI: 10.1103/PhysRevE.78.031912.
Conlon I, Raff M. 1999. Size control in animal development. Cell 96:235-244. DOI: 10.1016/s0092-8674(00)80563-2.
Jarrett AM, Lima EABF, Hormuth DA, McKenna MT, Feng X, Ekrut DA, Resende ACM, Brock A, Yankeelov TE. 2018. Mathematical models of tumor cell proliferation: A review of the literature. Expert Review of Anticancer Therapy 18:1271-1286. DOI: 10.1080/14737140.2018.1527689.
Mosca G, Adibi, M., Strauss, S., Runions, A., Sapala, A., Smith, R.S. 2018. Modeling Plant Tissue Growth and Cell Division. In Morris R., ed, Mathematical Modelling in Plant Biology. Springer, Cham.